Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Pharmacological Chaperones: The Time-Lapse
3. Pharmacological Chaperones: Rational and Application
4. Pharmacological Chaperones: Direct Specific Binding to Folded or Partially Folded Target Proteins as a Mechanistic Hallmark
5. Pharmacological Chaperones: The Quick Path to Success is not Always the Best One
6. Pharmacological Chaperones: Promising Drugs with a Restriction
7. Pharmacological Chaperones: Improvement of a Drug
8. Pharmacological Chaperones: Besides the Inhibitors
9. Pharmacological and Chemical Chaperones: Two Types of Drugs Often Confused
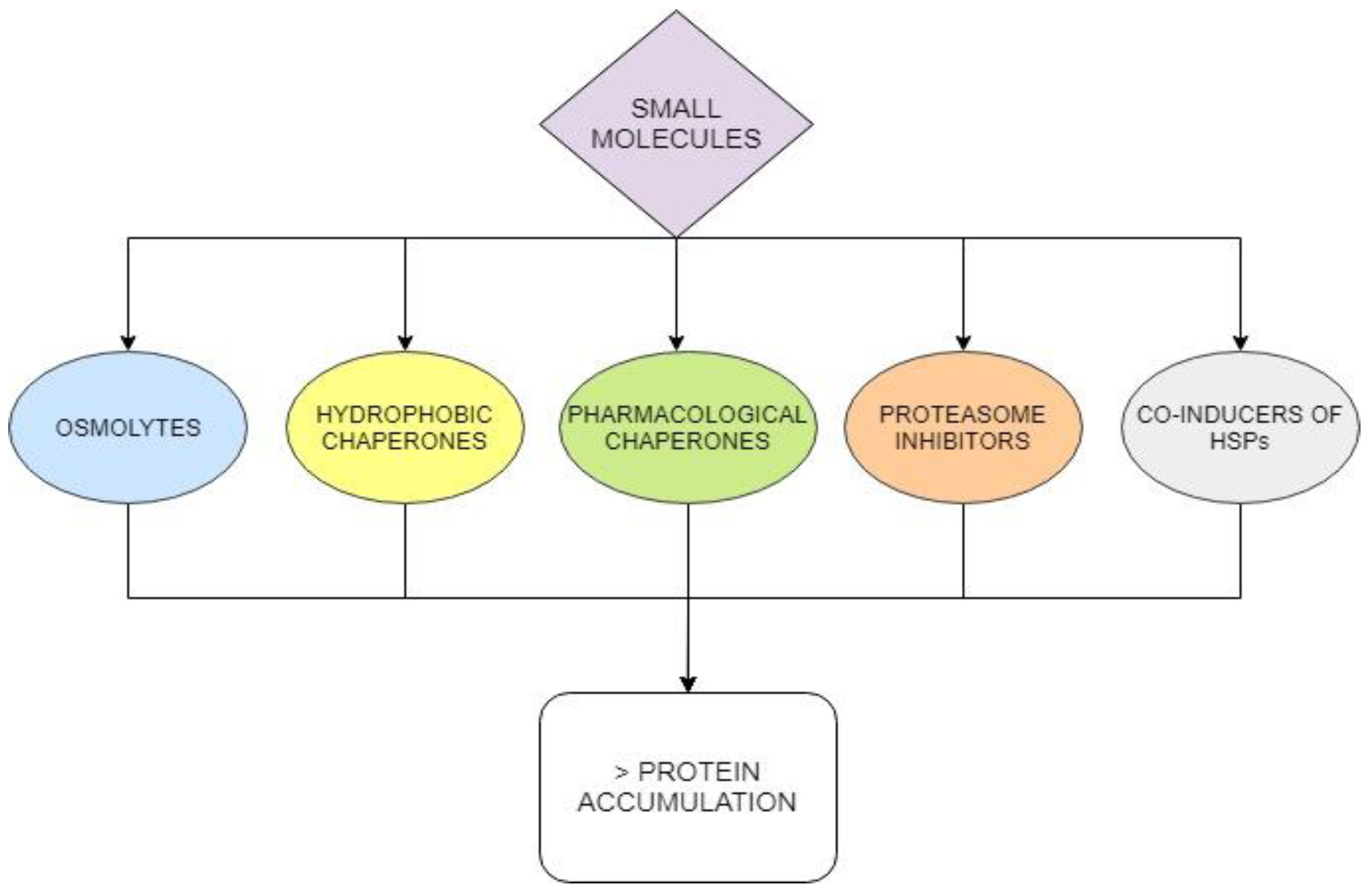
10. Other “Small Molecules”: Alternative or Synergistic Approaches to Treat Diseases Caused by Destabilizing Missense Mutations
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hou, Z.S.; Ulloa-Aguirre, A.; Tao, Y.X. Pharmacoperone drugs: Targeting misfolded proteins causing lysosomal storage- ion channels-, and g protein-coupled receptors-associated conformational disorders. Expert Rev. Clin. Pharmacol. 2018, 11, 611–624. [Google Scholar] [CrossRef]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Gámez, A.; Yuste-Checa, P.; Brasil, S.; Briso-Montiano, Á.; Desviat, L.R.; Ugarte, M.; Pérez-Cerdá, C.; Pérez, B. Protein misfolding diseases: Prospects of pharmacological treatment. Clin. Genet. 2018, 93, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Fernandez, E.M.; Garcia Fernandez, J.M.; Mellet, C.O. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from gaucher, gm1-gangliosidosis and fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matalonga, L.; Gort, L.; Ribes, A. Small molecules as therapeutic agents for inborn errors of metabolism. J. Inherit. Metab. Dis. 2017, 40, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Leidenheimer, N.J. Pharmacological chaperones: Beyond conformational disorders. In Targeting Trafficking in Drug Development; Springer: Cham, Switzerland, 2017; pp. 135–153. [Google Scholar]
- Shin, M.H.; Lim, H.-S. Screening methods for identifying pharmacological chaperones. Mol. Biosyst. 2017, 13, 638–647. [Google Scholar] [CrossRef]
- Betancor-Fernández, I.; Timson, D.J.; Salido, E.; Pey, A.L. Natural (and unnatural) small molecules as pharmacological chaperones and inhibitors in cancer. In Targeting Trafficking in Drug Development; Springer: Cham, Switzerland, 2017; pp. 155–190. [Google Scholar]
- Convertino, M.; Das, J.; Dokholyan, N.V. Pharmacological chaperones: Design and development of new therapeutic strategies for the treatment of conformational diseases. ACS Chem. Biol. 2016, 11, 1471–1489. [Google Scholar] [CrossRef]
- Tao, Y.X.; Conn, P.M. Chaperoning g protein-coupled receptors: From cell biology to therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar] [CrossRef] [Green Version]
- Aymami, J.; Barril, X.; Rodríguez-Pascau, L.; Martinell, M. Pharmacological chaperones for enzyme enhancement therapy in genetic diseases. Pharm. Pat. Anal. 2013, 2, 109–124. [Google Scholar] [CrossRef]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; García Fernández, J.M.; Mellet, C.O. Pharmacological chaperone therapy for gaucher disease: A patent review. Expert Opin. on Ther. Pat. 2011, 21, 885–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Morello, J.P.; Salahpour, A.; Laperriere, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petaja-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded v2 vasopressin receptor mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium, T. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Iny Stein, T.; Levitt, J.; Gershoni, M.; Morrey, C.P.; Safran, M.; Lancet, D. Malacards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef] [Green Version]
- Ringe, D.; Petsko, G.A. Q&a: What are pharmacological chaperones and why are they interesting? J. Biol. 2009, 8, 80. [Google Scholar]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
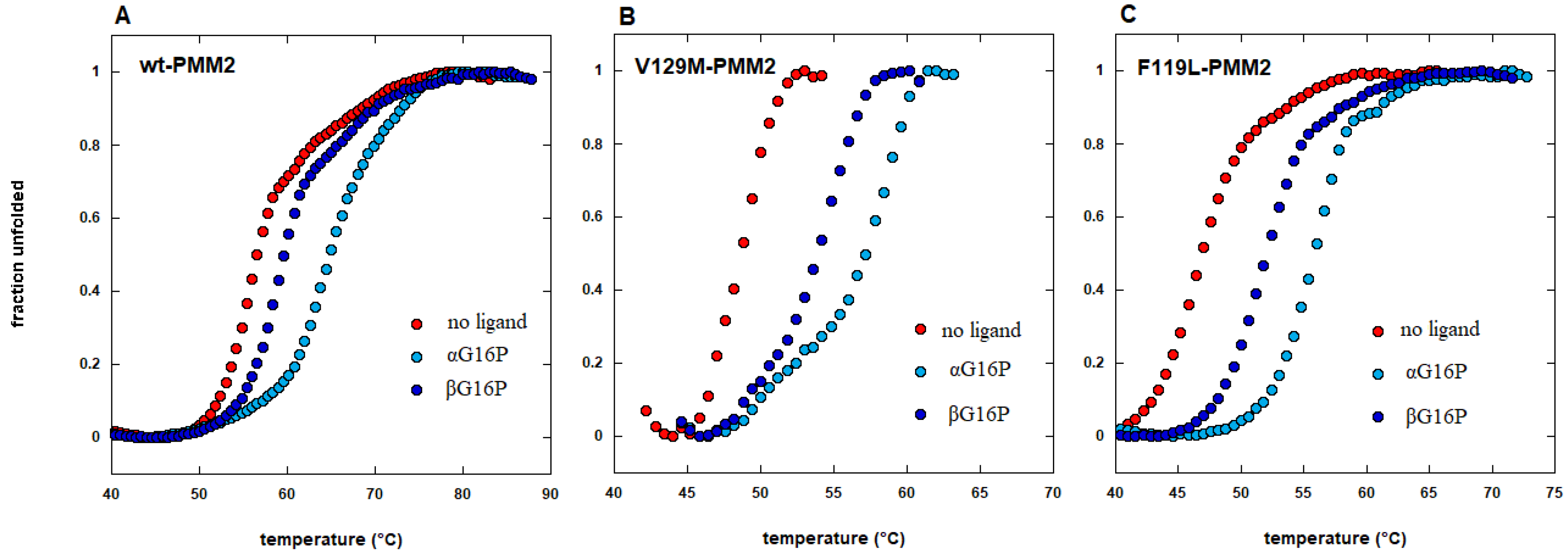
- Monticelli, M.; Liguori, L.; Allocca, M.; Andreotti, G.; Cubellis, M.V. Beta-glucose-1,6-bisphosphate stabilizes pathological phophomannomutase2 mutants in vitro and represents a lead compound to develop pharmacological chaperones for the most common disorder of glycosylation, pmm2-cdg. Int. J. Mol. Sci. 2019, 20, 4164. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, R.L.; D´Aquino, J.A.; Ringe, D.; Petsko, G.A. Effects of ph and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry 2009, 48, 4816–4827. [Google Scholar] [CrossRef] [Green Version]
- Guce, A.I.; Clark, N.E.; Rogich, J.J.; Garman, S.C. The molecular basis of pharmacological chaperoning in human alpha-galactosidase. Chem. Biol. 2011, 18, 1521–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreotti, G.; Citro, V.; Correra, A.; Cubellis, M.V. A thermodynamic assay to test pharmacological chaperones for fabry disease. Biochim. Biophys. Acta 2014, 1840, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein. Sci. 2015, 79, 28.9.1–28.9.14. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal alpha-galactosidase a in fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef]
- Sugawara, K.; Ohno, K.; Saito, S.; Sakuraba, H. Structural characterization of mutant α-galactosidases causing fabry disease. J. Hum. Genet. 2008, 53, 812. [Google Scholar] [CrossRef]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. The large phenotypic spectrum of fabry disease requires graduated diagnosis and personalized therapy: A meta-analysis can help to differentiate missense mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef] [Green Version]
- Okumiya, T.; Ishii, S.; Takenaka, T.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. Galactose stabilizes various missense mutants of alpha-galactosidase in fabry disease. Biochem. Biophys. Res. Commun. 1995, 214, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Chimenti, C.; Ricci, R.; Natale, L.; Russo, M.A.; Pieroni, M.; Eng, C.M.; Desnick, R.J. Improvement in cardiac function in the cardiac variant of fabry’s disease with galactose-infusion therapy. N. Engl. J. Med. 2001, 345, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Legler, G.; Pohl, S. Synthesis of 5-amino-5-deoxy-d-galactopyranose and 1,5-dideoxy-1,5-imino-d-galactitol, and their inhibition of alpha- and beta-d-galactosidases. Carbohydr. Res. 1986, 155, 119–129. [Google Scholar] [CrossRef]
- Bernotas, R.C.; Pezzone, M.A.; Ganem, B. Synthesis of (+)-1,5-dideoxy-1,5-imino-d-galactitol, a potent alpha-d-galactosidase inhibitor. Carbohydr. Res. 1987, 167, 305–311. [Google Scholar] [CrossRef]
- Tsukimura, T.; Chiba, Y.; Ohno, K.; Saito, S.; Tajima, Y.; Sakuraba, H. Molecular mechanism for stabilization of a mutant alpha-galactosidase a involving m51i amino acid substitution by imino sugars. Mol. Genet. Metab. 2011, 103, 26–32. [Google Scholar] [CrossRef]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of alpha-galactosidase a that respond to a pharmacological chaperone for fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef]
- Galafold. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/galafold (accessed on 9 June 2016).
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase a levels in fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The pharmacological chaperone isofagomine increases the activity of the gaucher disease l444p mutant form of beta-glucosidase. Febs. J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef]
- Moran, N. Fda approves galafold, a triumph for amicus. Nat. Biotechnol. 2018, 36, 913. [Google Scholar] [CrossRef]
- Nowak, A.; Uyen, H.D.; Krayenbuehl, P.A.; Beuschlein, F.; Schiffmann, R.; Barbey, F. Fabry disease genotype, phenotype and migalastat amenability: Insights from a national cohort. J. Inherit. Metab. Dis. 2019, 1–14. [Google Scholar] [CrossRef]
- Andreotti, G.; Monti, M.C.; Citro, V.; Cubellis, M.V. Heterodimerization of two pathological mutants enhances the activity of human phosphomannomutase2. PLoS ONE 2015, 10, e0139882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.J.; Rossi, B.; Tang, K.; Wu, X.; Mascioli, K.; Donaudy, F.; Tuzzi, M.R.; Fontana, F.; Cubellis, M.V.; Porto, C.; et al. The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum. Mutat. 2009, 30, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Estivill, X.; Casals, T.; Morral, N.; Chillon, M.; Bosch, A.; Nunes, V.; Gasparini, P.; Seia, A.; Pignatti, P.; Novelli, G. Δf508 gene deletion in cystic fibrosis in southern europe. Lancet 1989, 334, 1404. [Google Scholar] [CrossRef]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of cftr mutations—correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Gelbart, T.; Westwood, B.; Grabowski, G.; Beutler, E. High frequency of the gaucher disease mutation at nucleotide 1226 among ashkenazi jews. Am. J. Hum. Genet. 1991, 49, 855. [Google Scholar]
- Slatkin, M. A population-genetic test of founder effects and implications for ashkenazi jewish diseases. Am. J. Hum. Genet. 2004, 75, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Knospe, A.M.; Seemann, S.; Citro, V.; Cubellis, M.V.; Rolfs, A. In vitro enzyme measurement to test pharmacological chaperone responsiveness in fabry and pompe disease. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet 2013, 9, e1003632. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, G.; Citro, V.; De Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of fabry disease with pharmacological chaperones: From in silico predictions to in vitro tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Giugliani, R.; Waldek, S.; Germain, D.P.; Nicholls, K.; Bichet, D.G.; Simosky, J.K.; Bragat, A.C.; Castelli, J.P.; Benjamin, E.R.; Boudes, P.F. A phase 2 study of migalastat hydrochloride in females with fabry disease: Selection of population, safety and pharmacodynamic effects. Mol. Genet. Metab. 2013, 109, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Fabry_cep: A tool to identify fabry mutations responsive to pharmacological chaperones. Orphanet J. Rare Dis. 2013, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkinson, S.F.; Fleet, G.W.; Nash, R.J.; Koike, Y.; Adachi, I.; Yoshihara, A.; Morimoto, K.; Izumori, K.; Kato, A. Looking-glass synergistic pharmacological chaperones: Dgj and l-dgj from the enantiomers of tagatose. Org. Lett. 2011, 13, 4064–4067. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Kuanar, M.; Slavov, S.; Hall, C.D.; Karelson, M.; Kahn, I.; Dobchev, D.A. Quantitative correlation of physical and chemical properties with chemical structure: Utility for prediction. Chem. Rev. 2010, 110, 5714–5789. [Google Scholar] [CrossRef]
- Asano, N.; Ishii, S.; Kizu, H.; Ikeda, K.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.Q. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase a activity in fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef]
- Yu, Y.; Mena-Barragan, T.; Higaki, K.; Johnson, J.L.; Drury, J.E.; Lieberman, R.L.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular basis of 1-deoxygalactonojirimycin arylthiourea binding to human alpha-galactosidase a: Pharmacological chaperoning efficacy on fabry disease mutants. ACS Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Mena-Barragan, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.; Ohno, K.; Suzuki, Y.; Higaki, K.; Garcia Fernandez, J.M.; Ortiz Mellet, C. Ph-responsive pharmacological chaperones for rescuing mutant glycosidases. Angew. Chem. Int. Ed. Engl. 2015, 54, 11696–11700. [Google Scholar] [CrossRef]
- Lukas, J.; Pockrandt, A.M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Glaser, A.; Beller, M.; Rolfs, A.; et al. Enzyme enhancers for the treatment of fabry and pompe disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liou, B.; Xu, Y.H.; Quinn, B.; Zhang, W.; Hamler, R.; Setchell, K.D.; Grabowski, G.A. Ex vivo and in vivo effects of isofagomine on acid beta-glucosidase variants and substrate levels in gaucher disease. J. Biol. Chem. 2012, 287, 4275–4287. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, N.; Xu, Y.H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic gaucher disease: Dysregulated mrnas and mirnas in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef]
- Mena-Barragan, T.; Garcia-Moreno, M.I.; Sevsek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; Fernandez, J.M.G.; Mellet, C.O. Probing the inhibitor versus chaperone properties of sp(2)-iminosugars towards human beta-glucocerebrosidase: A picomolar chaperone for gaucher disease. Molecules 2018, 23, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citro, V.; Pena-Garcia, J.; den-Haan, H.; Perez-Sanchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an allosteric binding site on human lysosomal alpha-galactosidase opens the way to new pharmacological chaperones for fabry disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urquiza, P.; Lain, A.; Sanz-Parra, A.; Moreno, J.; Bernardo-Seisdedos, G.; Dubus, P.; Gonzalez, E.; Gutierrez-de-Juan, V.; Garcia, S.; Erana, H.; et al. Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria. Sci. Transl. Med. 2018, 10, eaat7467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, K.; Wen, A.; Cook, A.E.; Sexton, P.M.; Conigrave, A.D.; Christopoulos, A. Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 2013, 154, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Hotsubo, T.; Kobayashi, K.; Mochizuki, H.; Ishizu, K.; Tajima, T. Loss-of-function and gain-of-function mutations of calcium-sensing receptor: Functional analysis and the effect of allosteric modulators nps r-568 and nps 2143. J. Clin. Endocrinol. Metab. 2013, 98, E1692–E1701. [Google Scholar] [CrossRef] [Green Version]
- Newton, C.L.; Anderson, R.C. Pharmacoperones for misfolded gonadotropin receptors. Handb. Exp. Pharmacol. 2018, 245, 111–134. [Google Scholar]
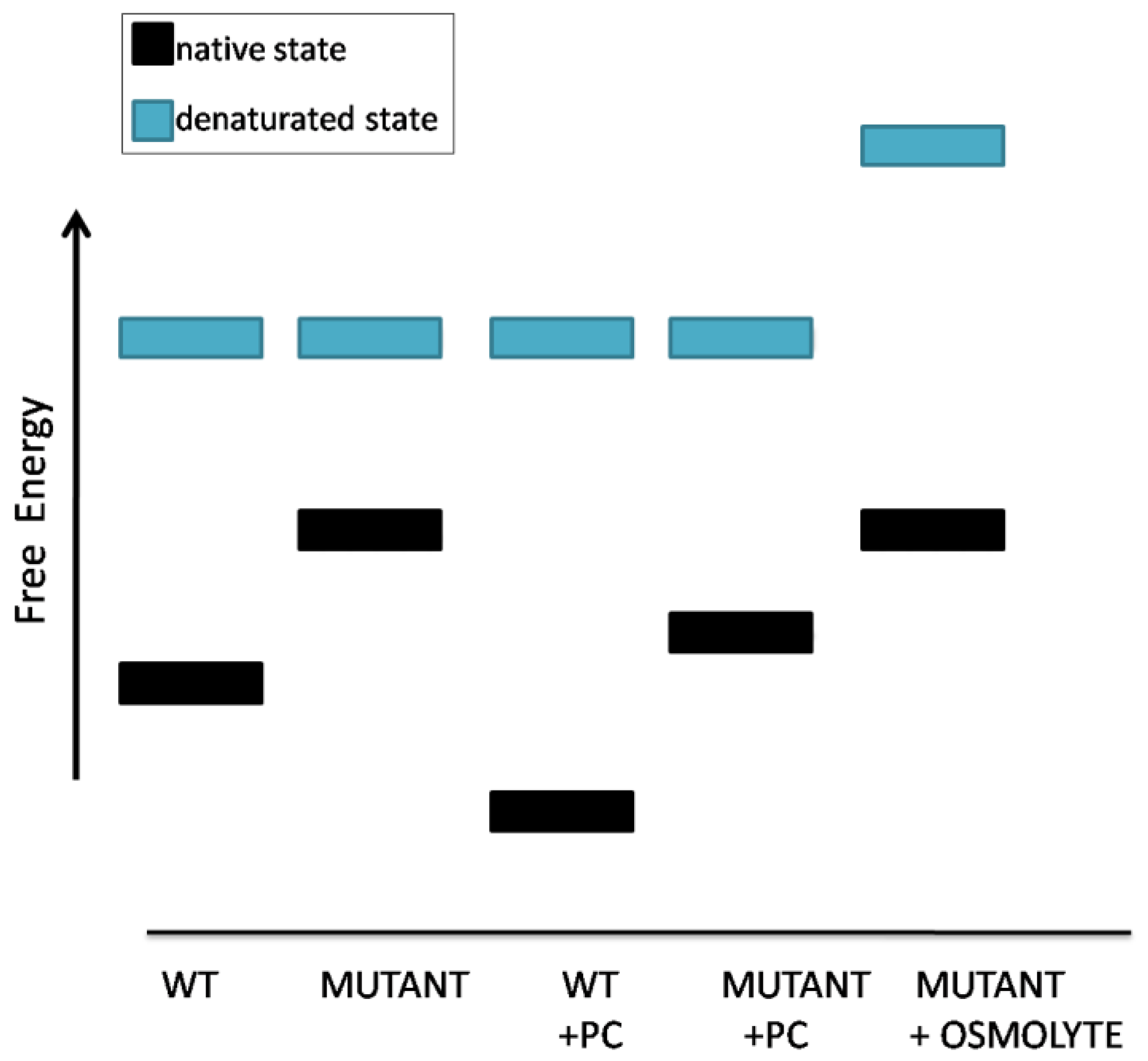
- Andreotti, G.; Pedone, E.; Giordano, A.; Cubellis, M.V. Biochemical phenotype of a common disease-causing mutation and a possible therapeutic approach for the phosphomannomutase 2-associated disorder of glycosylation. Mol. Genet. Genomic. Med. 2013, 1, 32–44. [Google Scholar] [CrossRef]
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef] [Green Version]
- Noorwez, S.M.; Malhotra, R.; McDowell, J.H.; Smith, K.A.; Krebs, M.P.; Kaushal, S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant p23h. J. Biol. Chem. 2004, 279, 16278–16284. [Google Scholar] [CrossRef] [Green Version]
- Kure, S.; Hou, D.C.; Ohura, T.; Iwamoto, H.; Suzuki, S.; Sugiyama, N.; Sakamoto, O.; Fujii, K.; Matsubara, Y.; Narisawa, K. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J. Pediatr. 1999, 135, 375–378. [Google Scholar] [CrossRef]
- Muntau, A.C.; Roschinger, W.; Habich, M.; Demmelmair, H.; Hoffmann, B.; Sommerhoff, C.P.; Roscher, A.A. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N. Engl. J. Med. 2002, 347, 2122–2132. [Google Scholar] [CrossRef]
- Bernegger, C.; Blau, N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: A study of 1,919 patients observed from 1988 to 2002. Mol. Genet. Metab. 2002, 77, 304–313. [Google Scholar] [CrossRef]
- Ismail, H.M.; Krishnamoorthy, N.; Al-Dewik, N.; Zayed, H.; Mohamed, N.A.; Giacomo, V.D.; Gupta, S.; Haberle, J.; Thony, B.; Blom, H.J.; et al. In silico and in vivo models for qatari-specific classical homocystinuria as basis for development of novel therapies. Hum. Mutat. 2019, 40, 230–240. [Google Scholar] [CrossRef]
- Banning, A.; Gulec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of small molecule compounds for pharmacological chaperone therapy of aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinting, S.; Hoppner, S.; Schindlbeck, U.; Forstner, M.E.; Harfst, J.; Wittmann, T.; Griese, M. Functional rescue of misfolding abca3 mutations by small molecular correctors. Hum. Mol. Genet. 2018, 27, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, J.A.; Willis, L.K.; Perlmutter, D.H. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-at) z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-at deficiency. Proc. Natl. Acad. Sci. USA 2000, 97, 1796–1801. [Google Scholar] [CrossRef] [Green Version]
- Choo-Kang, L.R.; Zeitlin, P.L. Induction of hsp70 promotes deltaf508 cftr trafficking. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2001, 281, L58–L68. [Google Scholar] [CrossRef]
- Le Saux, O.; Fulop, K.; Yamaguchi, Y.; Ilias, A.; Szabo, Z.; Brampton, C.N.; Pomozi, V.; Huszar, K.; Aranyi, T.; Varadi, A. Expression and in vivo rescue of human abcc6 disease-causing mutants in mouse liver. PLoS ONE 2011, 6, e24738. [Google Scholar] [CrossRef] [Green Version]
- Sorrenson, B.; Suetani, R.J.; Williams, M.J.; Bickley, V.M.; George, P.M.; Jones, G.T.; McCormick, S.P. Functional rescue of mutant abca1 proteins by sodium 4-phenylbutyrate. J. Lipid Res. 2013, 54, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Cinque, L.; Sparaneo, A.; Penta, L.; Mencarelli, A.; Rogaia, D.; Esposito, S.; Fabrizio, F.P.; Baorda, F.; Verrotti, A.; Falorni, A.; et al. Autosomal dominant pth gene signal sequence mutation in a family with familial isolated hypoparathyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, P.V.; Stapelbroek, J.M.; Krieger, E.; de Bie, P.; van de Graaf, S.F.; de Groot, R.E.; van Beurden, E.; Spijker, E.; Houwen, R.H.; Berger, R. Reduced expression of atp7b affected by wilson disease–causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology 2009, 50, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Lyons, B.M. Sodium 4-phenylbutyrate downregulates hsc70 expression by facilitating mrna degradation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L43–L51. [Google Scholar] [CrossRef] [PubMed]
- Suaud, L.; Miller, K.; Panichelli, A.E.; Randell, R.L.; Marando, C.M.; Rubenstein, R.C. 4-phenylbutyrate stimulates hsp70 expression through the elp2 component of elongator and stat-3 in cystic fibrosis epithelial cells. J. Biol. Chem. 2011, 286, 45083–45092. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.S.; Zeng, L.R.; Quan, R.F.; Tang, Y.H.; Zheng, W.J.; Qu, G.; Xu, C.D.; Zhu, F.B.; Huang, Z.M. 4-phenylbutyrate protects rat skin flaps against ischemia-reperfusion injury and apoptosis by inhibiting endoplasmic reticulum stress. Mol. Med. Rep. 2016, 13, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef]
- Cousens, L.S.; Gallwitz, D.; Alberts, B.M. Different accessibilities in chromatin to histone acetylase. J. Biol. Chem. 1979, 254, 1716–1723. [Google Scholar]
- Mimori, S.; Ohtaka, H.; Koshikawa, Y.; Kawada, K.; Kaneko, M.; Okuma, Y.; Nomura, Y.; Murakami, Y.; Hamana, H. 4-phenylbutyric acid protects against neuronal cell death by primarily acting as a chemical chaperone rather than histone deacetylase inhibitor. Bioorganic Med. Chem. Lett. 2013, 23, 6015–6018. [Google Scholar] [CrossRef]
- McDermott, C.J. Clinical trials in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2019, 32, 758–763. [Google Scholar] [CrossRef]
- Fog, C.K.; Zago, P.; Malini, E.; Solanko, L.M.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.H.; Bembi, B. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Parfitt, D.; Aguila, M.; McCulley, C.; Bevilacqua, D.; Mendes, H.; Athanasiou, D.; Novoselov, S.; Kanuga, N.; Munro, P.; Coffey, P. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef] [Green Version]
- Mu, T.-W.; Ong, D.S.T.; Wang, Y.-J.; Balch, W.E.; Yates, J.R.; Segatori, L.; Kelly, J.W. Proteostasis regulators and pharmacologic chaperones synergize to correct protein misfolding diseases. Cell 2008, 134, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.M.; Brown, I.R. Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones 2007, 12, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the chinese “thunder of god vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawkar, A.R.; Cheng, W.C.; Beutler, E.; Wong, C.H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of n370s β-glucosidase: A therapeutic strategy for gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.M.; Haake, L.R.; Citro, V.; Brauer, A.U.; et al. Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in fabry disease. Biochem. J. 2020, 477. [Google Scholar] [CrossRef]








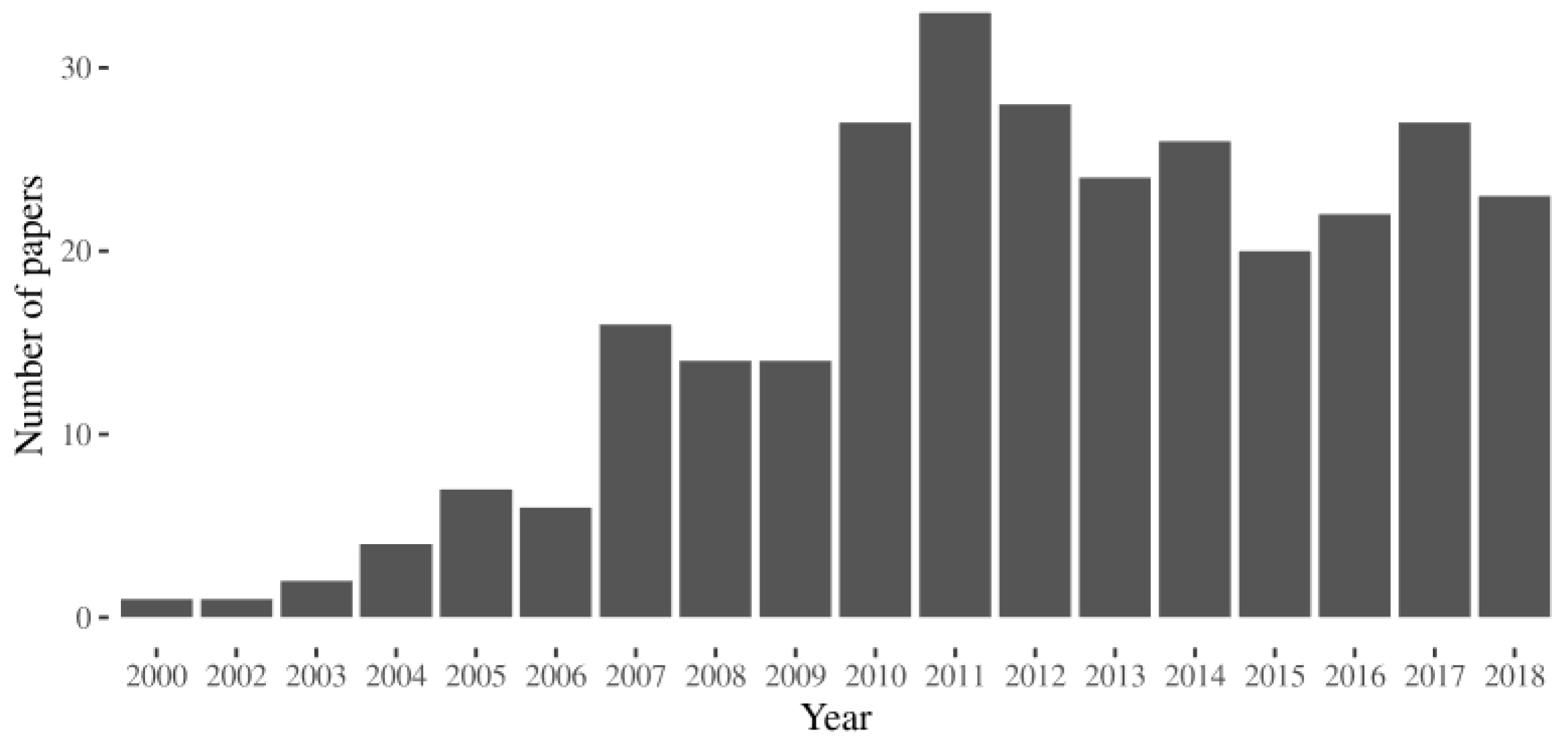{kind=link}
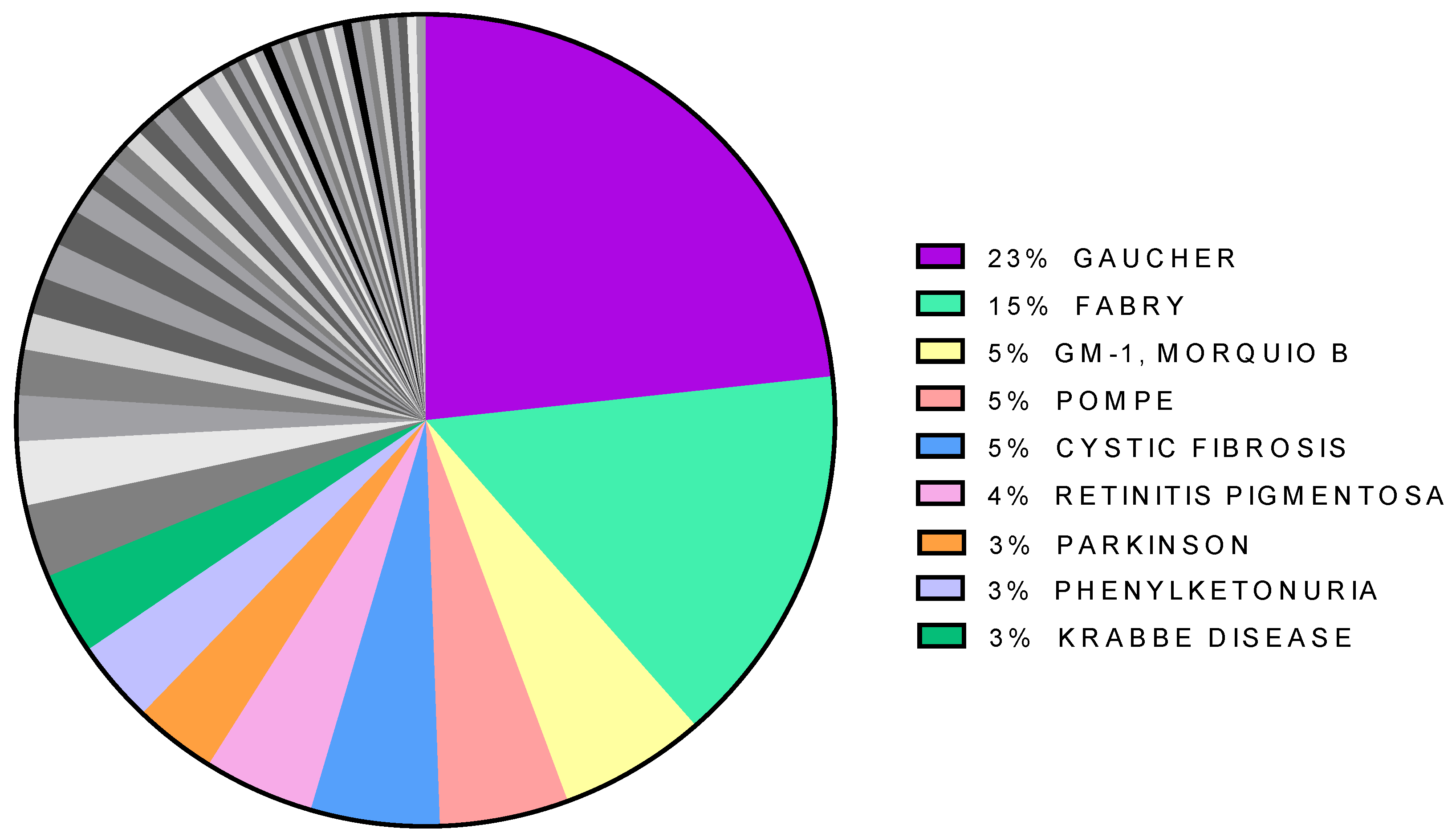{kind=link}
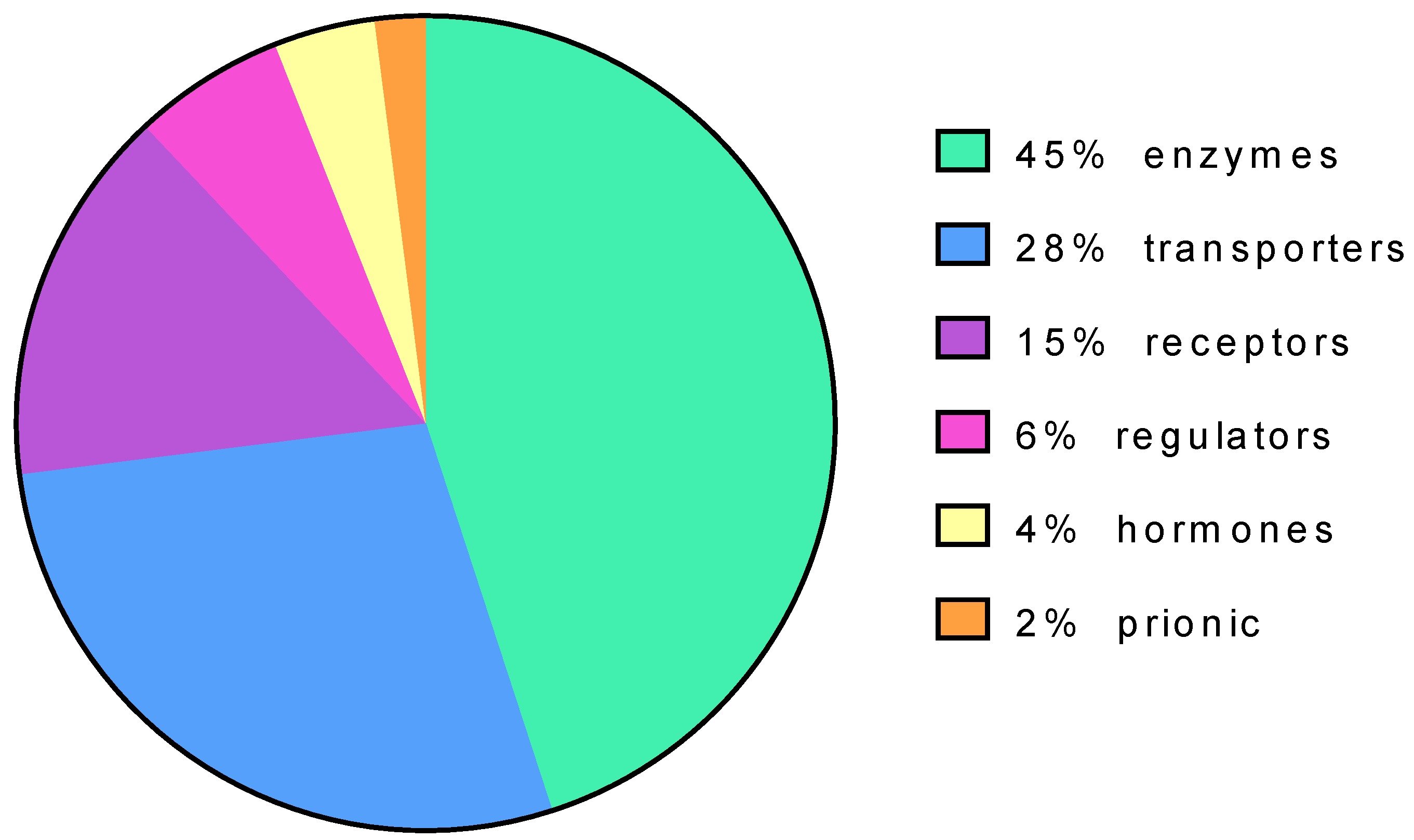{kind=link}
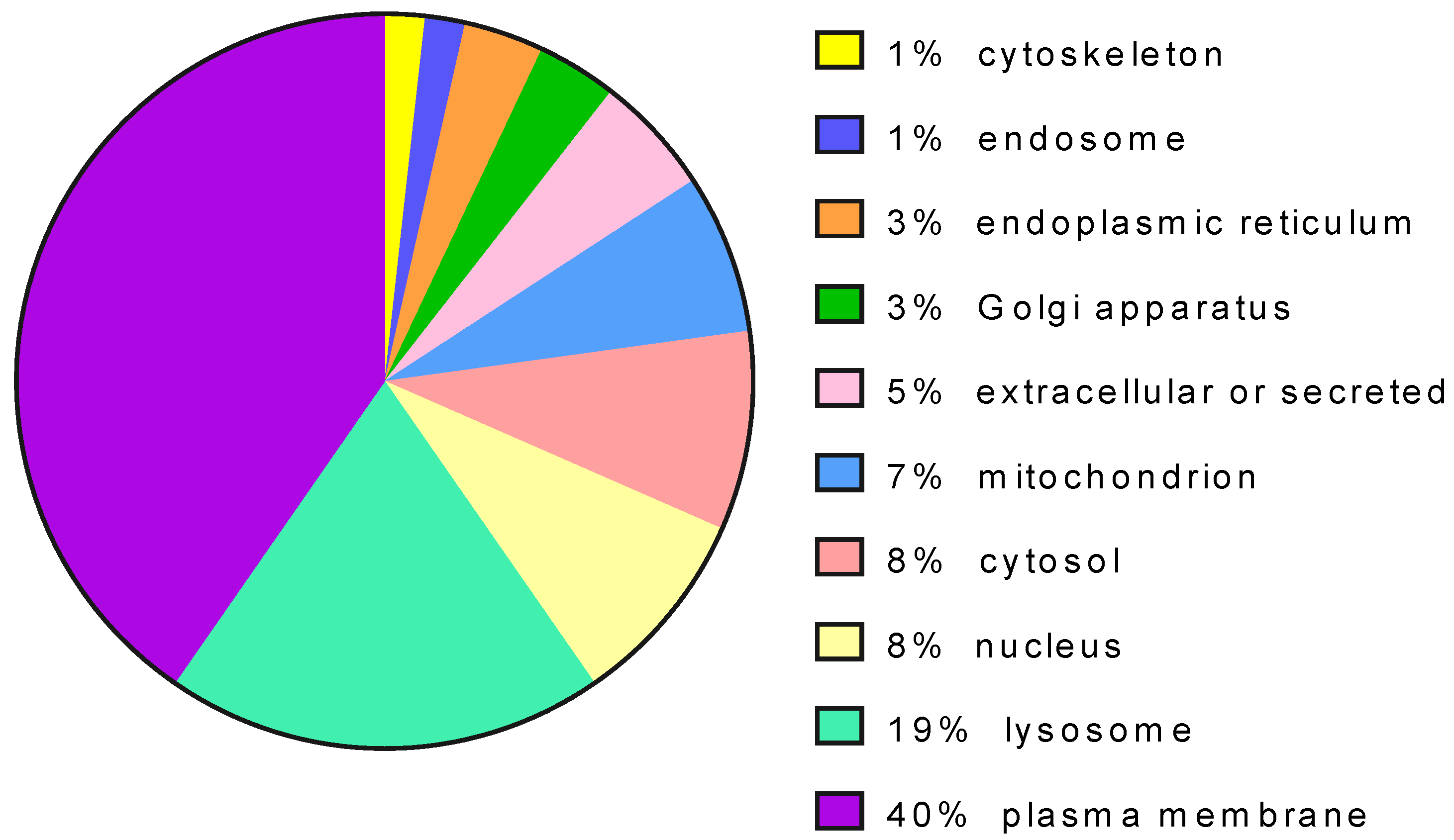{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Uniprot Code | Protein Type | Subcellular Location | N. Of Articles |
|---|---|---|---|---|---|
| Gaucher | GBA | P04062 | enzyme | lysosome | 64 |
| Fabry | GLA | P06280 | enzyme | lysosome | 42 |
| GM-1, Morqio B | GLB1 | P16278 | enzyme | lysosome | 16 |
| Pompe | GAA | P10253 | enzyme | lysosome | 14 |
| Cystic fibrosis | CFTR | P13569 | transporter | plasma membrane | 14 |
| Retinitis pigmentosa | RHO | P08100 | receptor | plasma membrane | 12 |
| Phenylketonuria | PAH | P00439 | enzyme | cytosol | 9 |
| Krabbe disease | GALC | P54803 | enzyme | lysosome | 9 |
| Nephrogenic diabetes insipidus | V2R | P30518 | receptor | plasma membrane | 8 |
| Long QT Syndrome | KCNH2 | Q12809 | transporter | plasma membrane | 7 |
| Parkinson | PARK7 | Q99497 | enzyme | plasma membrane, nucleus, mitochondrion | 5 |
| Niemann-Pick | NPC1 | O15118 | receptor | lysosome | 5 |
| Hyperoxaluria | AGXT | Q86XE5 | enzyme | mitochondrion | 5 |
| Obesity | MC4R | P32245 | receptor | plasma membrane | 4 |
| GM-2, Sanfilippo syndrome | GNRHR | P07686 | enzyme | lysosome | 4 |
| GM-2, Tay-Sachs syndrome | HEXB | P06865 | enzyme | lysosome | 4 |
| Galactosemia | HEXA | P07902 | enzyme | cytosol | 4 |
| Hypoparathyroidism | PTH | P01270 | hormone | extracellular or secreted | 3 |
| Parkinson | GALT | P04062 | enzyme | lysosome | 3 |
| Hypogonadotropic hypogonadism | ATP7B | P30968 | receptor | plasma membrane | 2 |
| Wilson | PMM2 | P35670 | transporter | Golgi apparatus | 2 |
| PMM2-CDG | SLC26A4 | O15305 | enzyme | cytosol | 2 |
| Pendred | MMAB | O43511 | transporter | plasma membrane | 2 |
| Methylmalonic aciduria | ABCB4 | Q96EY8 | enzyme | mitochondrion | 2 |
| Intrahepatic cholestasis | DRD4 | P21439 | transporter | plasma membrane | 2 |
| Hyperactivity disorder | ABCC8 | P21917 | receptor | plasma membrane | 2 |
| Diabetes | GPR56 | Q09428 | receptor | plasma membrane | 2 |
| Polymicrogyria | PGK1 | Q9Y653 | receptor | plasma membrane, extracellular or secreted | 1 |
| Phosphoglycerate kinase 1 deficiency | SNCA | P00558 | enzyme | cytosol | 1 |
| Parkinson | SUMF1 | P37840 | regulator | presynaptic vesicle | 1 |
| Multiple sulfatase deficiency | NPM | Q8NBK3 | enzyme | E.R. | 1 |
| Leukemia | PKR2 | P06748 | regulator | nucleus, cytoskeleton | 1 |
| Intrahepatic cholestasis | ABCB11 | O95342 | transporter | plasma membrane | 1 |
| Nocturnal frontal lobe epilepsy | CHRNB2/CHRNA4 | P17787/P43681 | transporter | plasma membrane | 1 |
| Hypomagnesemia | CLDN16 | Q9Y5I7 | transporter | plasma membrane | 1 |
| Creutzfeld-Jacob, Kuru | PRNP | P04156 | unclear/prion | plasma membrane | 1 |
| Homocystinuria | CBS | P35520 | enzyme | nucleus | 1 |
| Fibrodysplasia ossificans | ACVR1 | Q04771 | enzyme | plasma membrane | 1 |
| Epilepsy, Migraine | SCN1A | P35498 | transporter | plasma membrane | 1 |
| Dystonia | SLC2A1 | P11166 | transporter | plasma membrane | 1 |
| Diarrhea (cholera toxin) | NHE3 | P48764 | transporter | plasma membrane | 1 |
| Diabetes | KCNJ11 | Q14654 | transporter | plasma membrane | 1 |
| Intrahepatic cholestasis | ATP8B1 | O43520 | transporter | plasma membrane, Golgi apparatus, E.R. | 1 |
| Breast cancer | NBS | O60934 | regulator | nucleus | 1 |
| Amyotrophic lateral sclerosis | SOD1 | P00441 | enzyme | nucleus, mitochondrion | 1 |
| Amyloidosis | VPS29/VPS26 | Q9UBQ0/O75436 | transporter | endosome | 1 |
| Allan-Herndon-Dudley | SLC16A2 | P36021 | transporter | plasma membrane | 1 |
| Alkaptonuria | HGD | Q93099 | enzyme | cytosol | 1 |
| Aspartylglucosaminuria | AGA | P20933 | enzyme | lysosome | 1 |
| Ceroid lipofuscinosis | PPT1 | P50897 | enzyme | lysosome | 1 |
| Schindler disease | NAGA | P17050 | enzyme | lysosome | 1 |
| Diabetes mellitus | IAPP | P10997 | hormone | extracellular or secreted | 1 |
| GM-1 | IDS | P22304 | enzyme | lysosome | 1 |
| Morquio A, Hunter disease | GALNS | P34059 | enzyme | lysosome | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. https://doi.org/10.3390/ijms21020489
Liguori L, Monticelli M, Allocca M, Hay Mele B, Lukas J, Cubellis MV, Andreotti G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. International Journal of Molecular Sciences. 2020; 21(2):489. https://doi.org/10.3390/ijms21020489
Chicago/Turabian StyleLiguori, Ludovica, Maria Monticelli, Mariateresa Allocca, Bruno Hay Mele, Jan Lukas, Maria Vittoria Cubellis, and Giuseppina Andreotti. 2020. "Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations" International Journal of Molecular Sciences 21, no. 2: 489. https://doi.org/10.3390/ijms21020489
APA StyleLiguori, L., Monticelli, M., Allocca, M., Hay Mele, B., Lukas, J., Cubellis, M. V., & Andreotti, G. (2020). Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. International Journal of Molecular Sciences, 21(2), 489. https://doi.org/10.3390/ijms21020489