Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
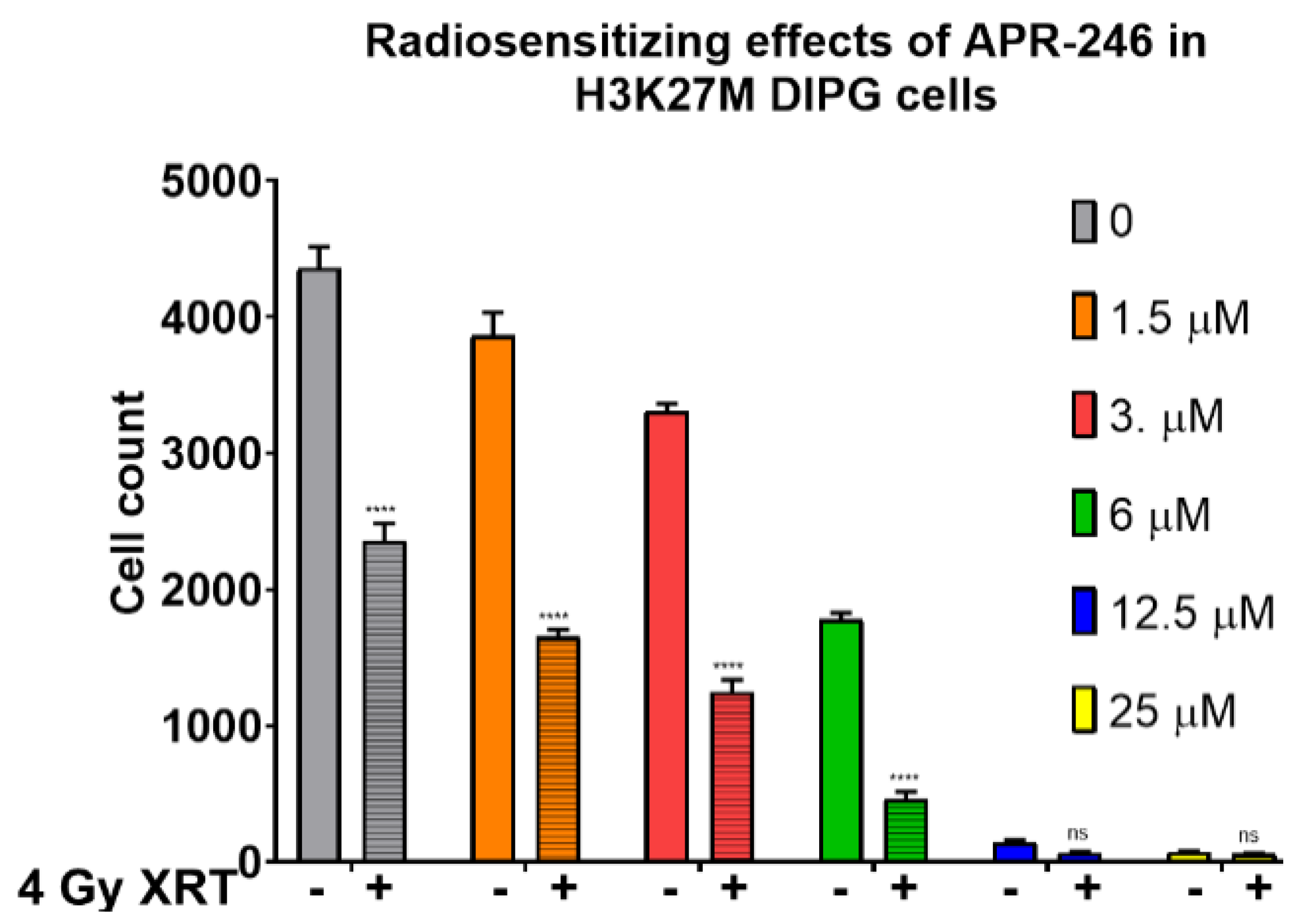
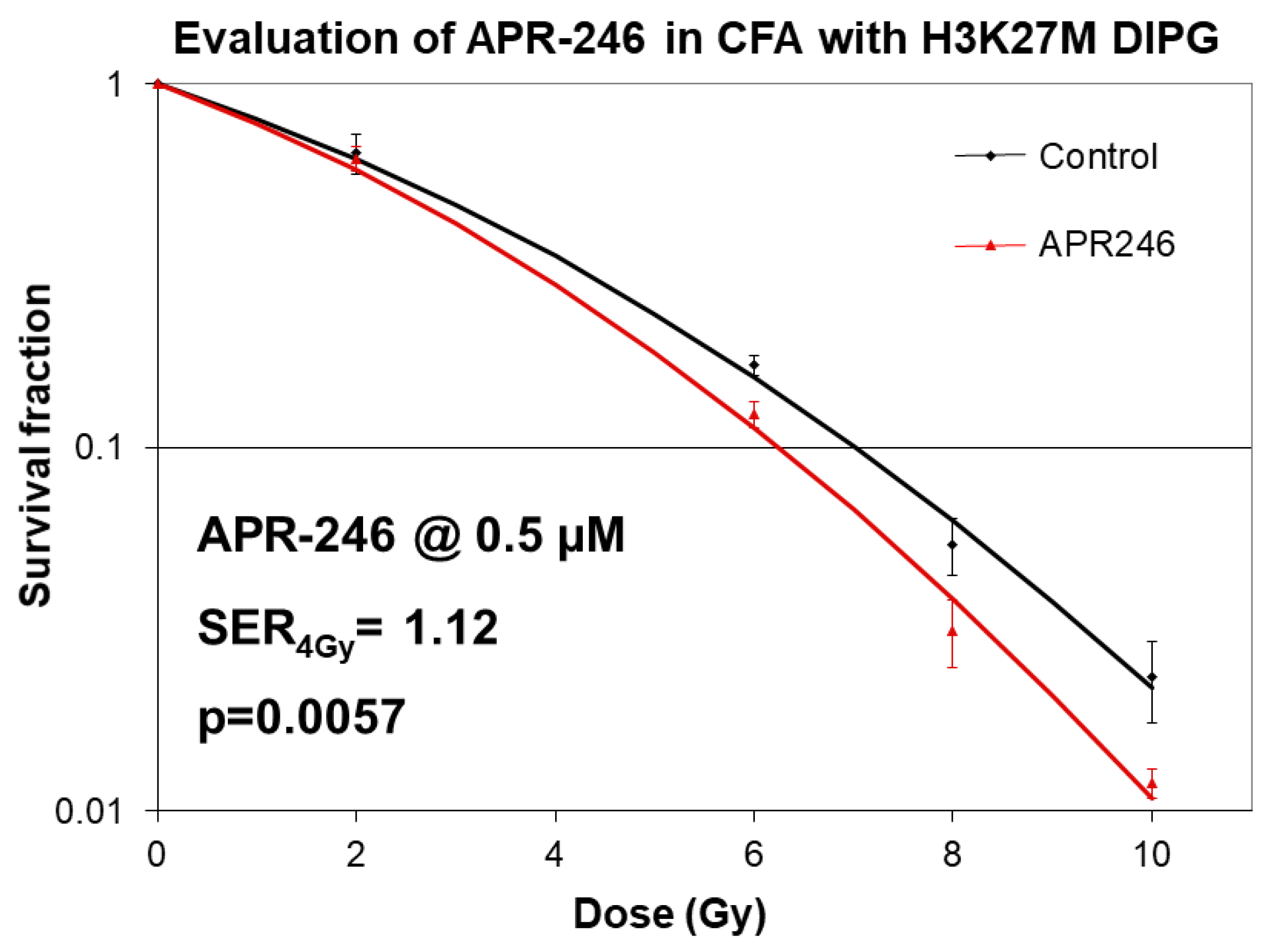
2.1. Mutant p53 Targeting Elicits Growth Inhibitory and Radio-Sensitizing Effects in H3K27M DIPG Cells
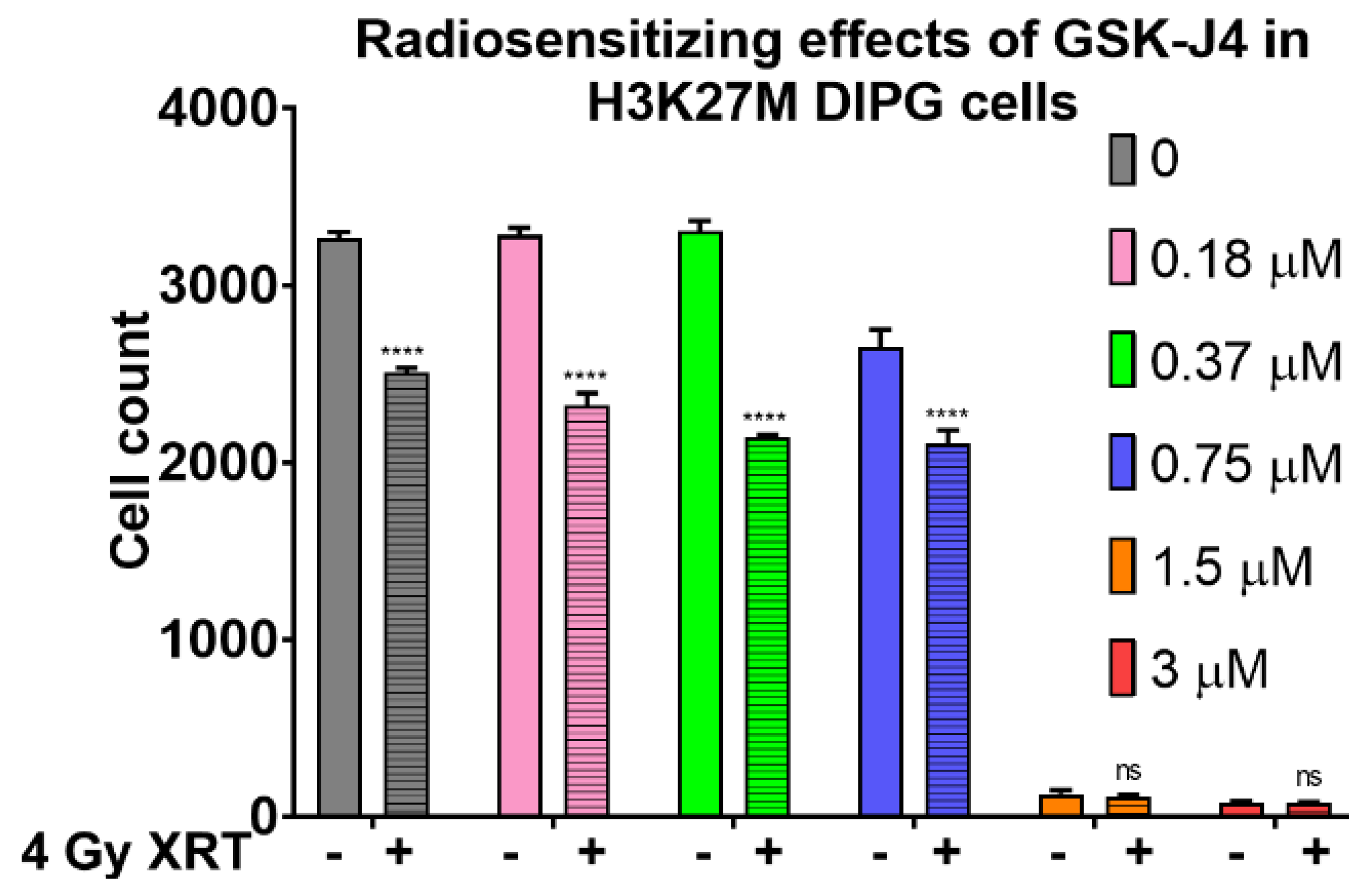
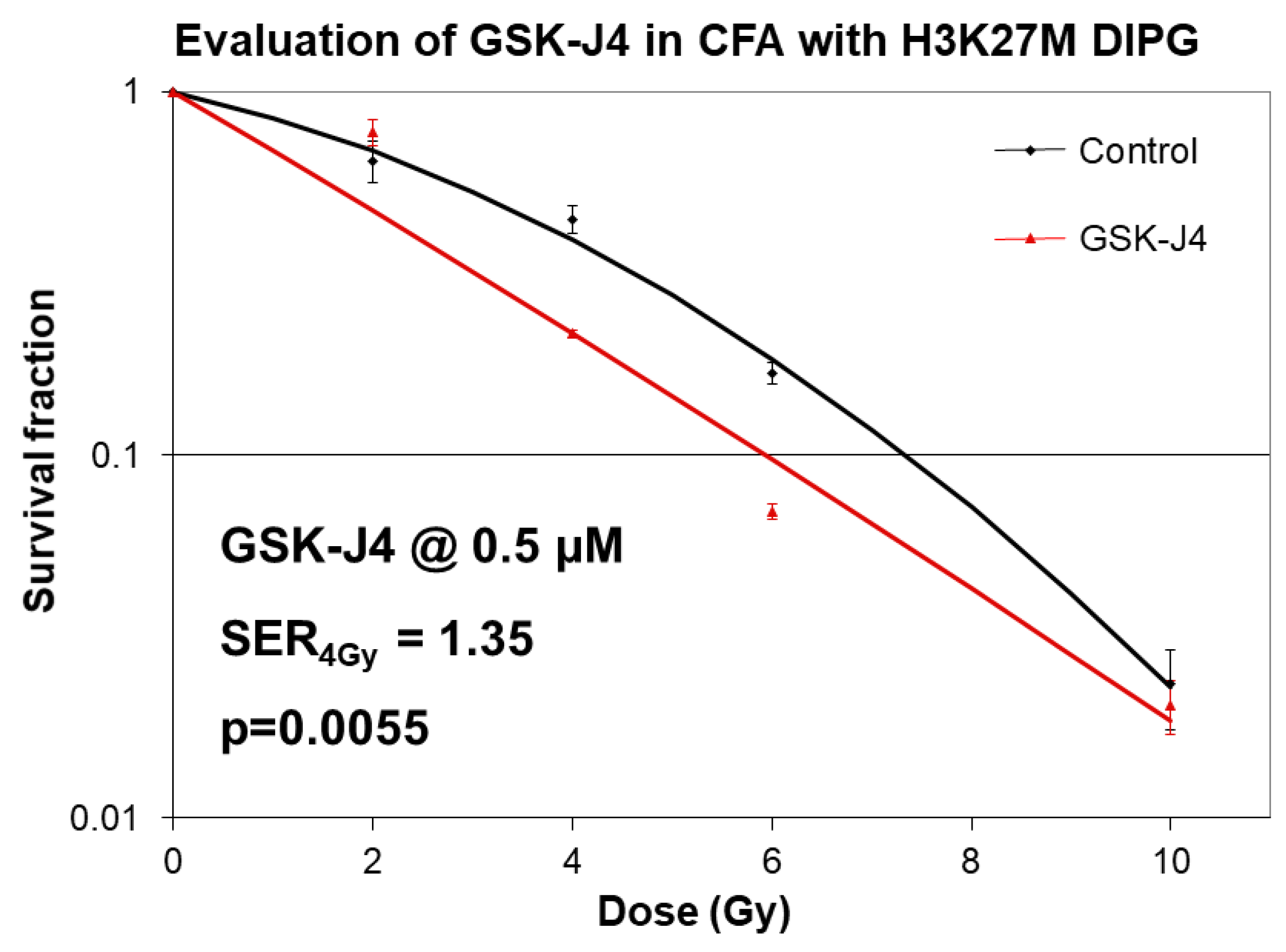
2.2. Inhibition of Jumonji Family Histone Demethylase with GSK-J4 Displays Anti-Proliferative and Radiosensitizing Effects on H3K27M Diffuse Intrinsic Pontine Glioma (DIPG) Cells
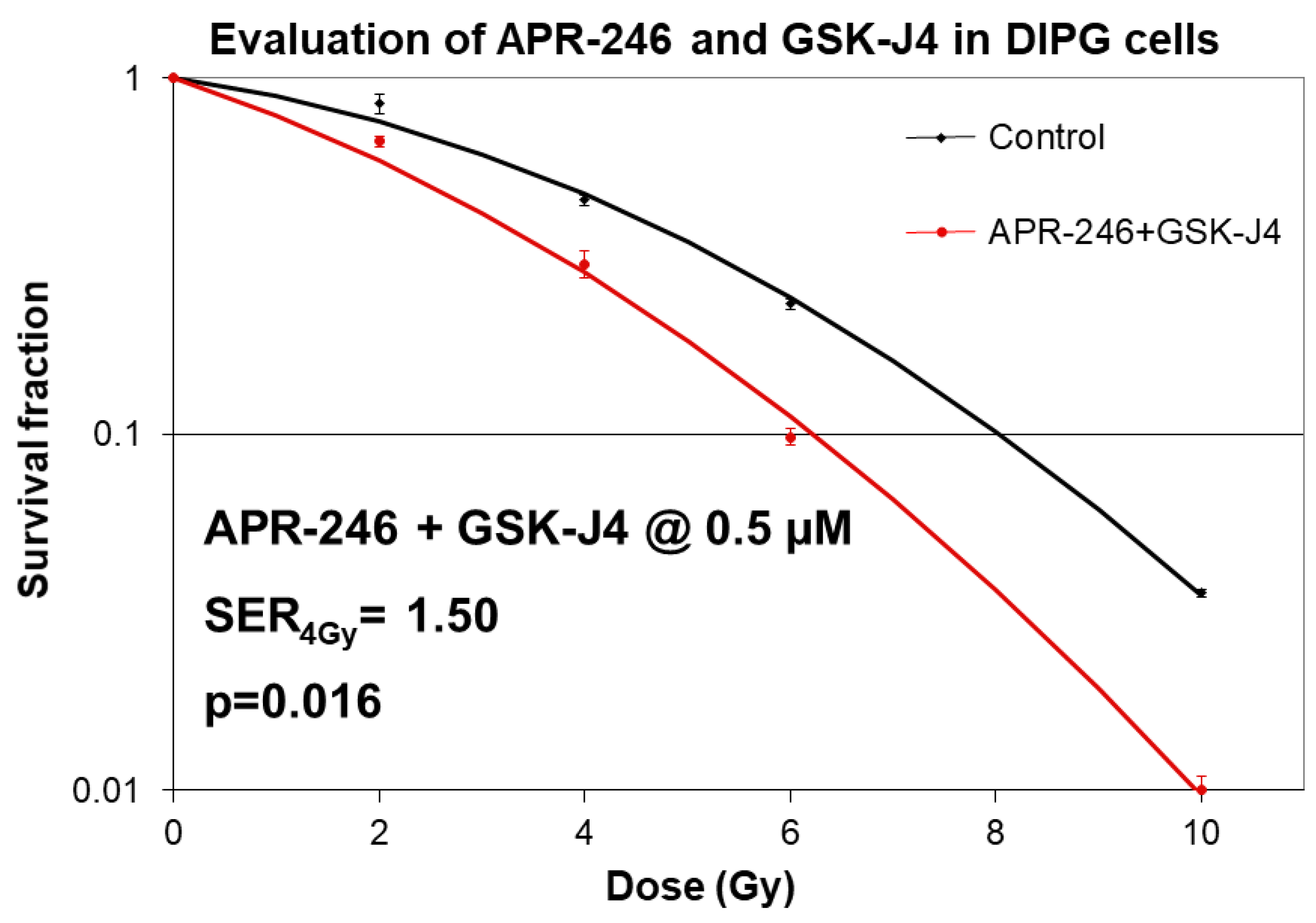
2.3. Combined Targeting of Mutant p53 and Jumonji Family Histone Improves Sensitivity of H3K27M DIPG Cells to Radiation
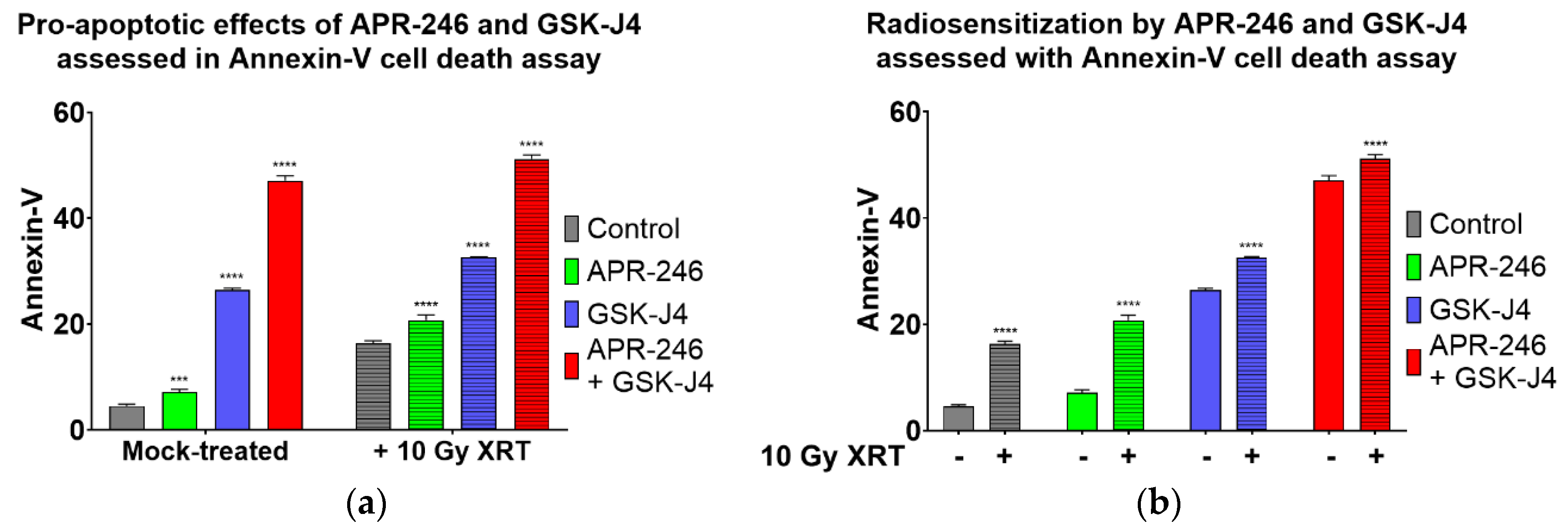
2.4. Combined Inhibition of Jumonji Family Histone Demethylase with Mutant p53 Targeting and Oxidative Stress Induction Promotes Apoptosis of H3K27M DIPG Cells
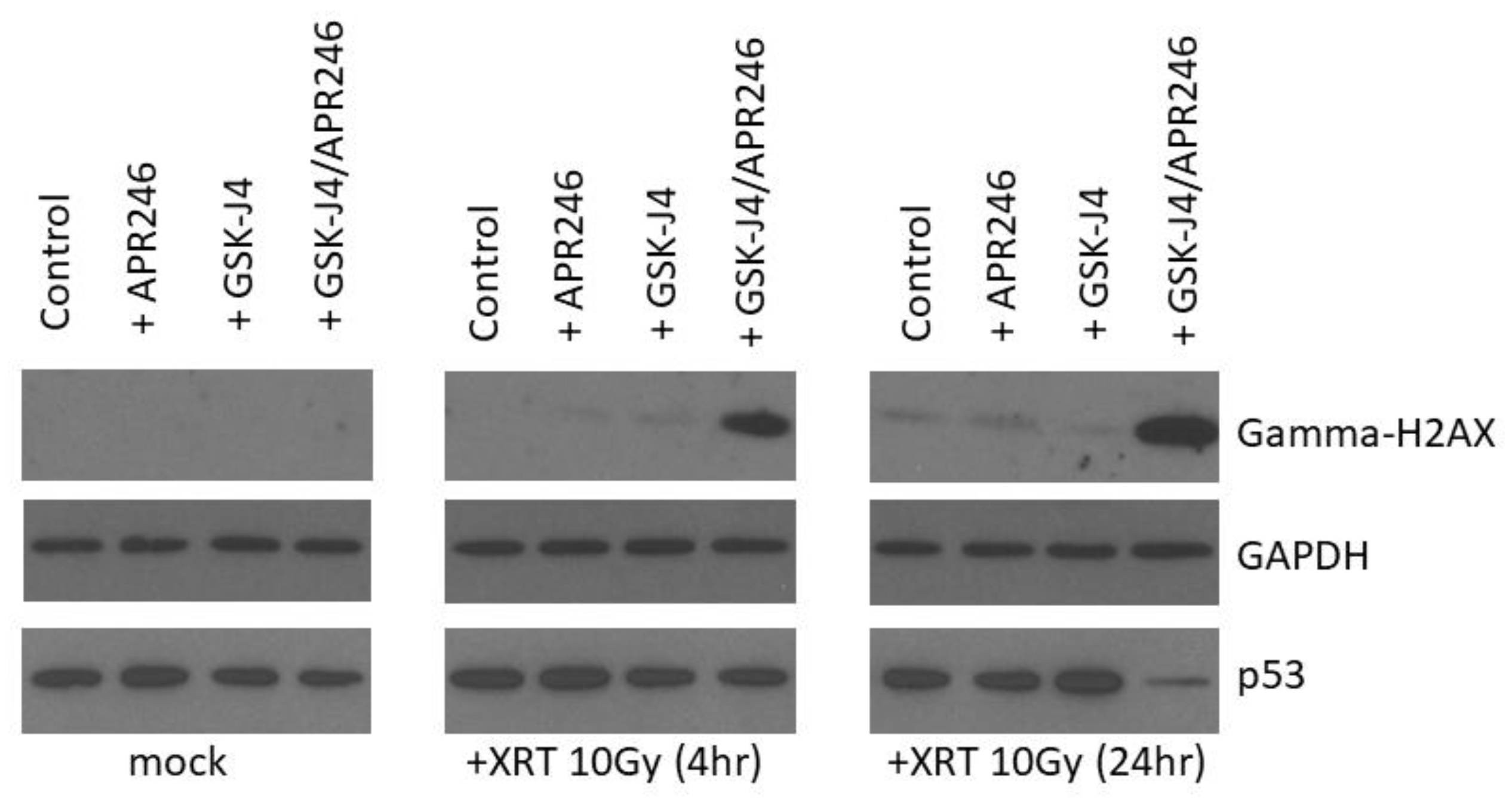
2.5. Combined Targeting of H3K27M and Mutant p53 Driven Disease Mechanisms Induces a DNA Damage Repair Deficit Following Irradiation
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| H3K27M | Histone H3 Lysine 27 to Methionine Mutation |
| DIPG | Diffuse Intrinsic Pontine Glioma |
| DSBs | Double Strand Breaks |
| CD | Calculated Dose |
| XRT | Ionizing radiation treatment |
| APR-246 | Mutant p53-reactivating agent |
| GSK-J4 | Jumonji family H3K27 histone demethylase inhibitor |
| SER | Sensitizer Enhancement Ratio |
| ROS | Reactive oxygen species |
References
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimo, P.; Panandiker, A.S.P.; Thompson, C.J.; Boop, F.A.; Qaddoumi, I.; Gajjar, A.; Armstrong, G.T.; Ellison, D.W.; Kun, L.E.; Ogg, R.J.; et al. Management and outcome of focal low-grade brainstem tumors in pediatric patients: The St. Jude experience. J. Neurosurg. Pediatr. 2013, 11, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.K.; Brandon, J.; Foreman, N.K.; Fenton, L.Z. Conventional MRI at presentation does not predict clinical response to radiation therapy in children with diffuse pontine glioma. Pediatr. Radiol. 2009, 39, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.A.; Liu, J.; Kieran, M.; Jabado, N.; Picton, S.; Packer, R.; St Rose, C.; Van Meeteren, A.S.; Carvalho, A.; et al.; CPNParis 2011 Conference Consensus, Group A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol. 2013, 15, 462–468. [Google Scholar] [CrossRef]
- Frazier, J.L.; Lee, J.; Thomale, U.W.; Noggle, J.C.; Cohen, K.J.; Jallo, G.I. Treatment of diffuse intrinsic brainstem gliomas: Failed approaches and future strategies. J. Neurosurg. Pediatr. 2009, 3, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Wolff, J.E.A.; Westphal, S.; Mölenkamp, G.; Gnekow, A.; Warmuth-Metz, M.; Rating, D.; Kuehl, J. Treatment of paediatric pontine glioma with oral trophosphamide and etoposide. Br. J. Cancer 2002, 87, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Korones, D.N.; Fisher, P.G.; Kretschmar, C.; Zhou, T.; Chen, Z.; Kepner, J.; Freeman, C. Treatment of children with diffuse intrinsic brain stem glioma with radiotherapy, vincristine and oral VP-16: A Children’s Oncology Group phase II study. Pediatr. Blood Cancer 2008, 50, 227–230. [Google Scholar] [CrossRef]
- Cohen, K.J.; Heideman, R.L.; Zhou, T.; Holmes, E.J.; Lavey, R.S.; Bouffet, E.; Pollack, I.F. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: A report from the Children’s Oncology Group. Neuro Oncol. 2011, 13, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Jennings, M.T.; Sposto, R.; Boyett, J.M.; Vezina, L.G.; Holmes, E.; Berger, M.S.; Bruggers, C.S.; Bruner, J.M.; Chan, K.W.; Dusenbery, K.E.; et al. Preradiation chemotherapy in primary high-risk brainstem tumors: Phase II study CCG-9941 of the Children’s Cancer Group. J. Clin. Oncol. 2002, 20, 3431–3437. [Google Scholar] [CrossRef]
- Mandell, L.R.; Kadota, R.; Freeman, C.; Douglass, E.C.; Fontanesi, J.; Cohen, M.E.; Kovnar, E.; Burger, P.; Sanford, R.A.; Kepner, J.; et al. There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: Results of a Pediatric Oncology Group phase III trial comparing conventional vs. hyperfractionated radiotherapy. Int. J. Radiat Oncol. Biol. Phys. 1999, 43, 959–964. [Google Scholar] [CrossRef]
- Janssens, G.O.; Jansen, M.H.; Lauwers, S.J.; Nowak, P.J.; Oldenburger, F.R.; Bouffet, E.; Saran, F.; Kamphuis-van Ulzen, K.; van Lindert, E.J.; Schieving, J.H.; et al. Hypofractionation vs conventional radiation therapy for newly diagnosed diffuse intrinsic pontine glioma: A matched-cohort analysis. Int. J. Radiat Oncol. Biol. Phys. 2013, 85, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, M.S.; Eldebawy, E.; Ahmed, S.; Mousa, A.G.; Amin, A.; Refaat, A.; Zaky, I.; Elkhateeb, N.; Sabry, M. Hypofractionated conformal radiotherapy for pediatric diffuse intrinsic pontine glioma (DIPG): A randomized controlled trial. Radiother. Oncol. 2014, 111, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hawkins, C. Pathology, Molecular Genetics and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 30, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Bykov, V.J.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Steensma, D.P.; Sweet, K.L.; Cluzeau, T.; Sekeres, M.A.; Garcia-Manero, G.; Roboz, G.J.; McLemore, A.F.; McGraw, K.L.; et al. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood 2018, 132, 3091. [Google Scholar] [CrossRef]
- Aprea Therapeutics. Receives FDA Fast Track Designation and Orphan Drug Designation for APR-246 for the Treatment of Myelodysplastic Syndromes (MDS) [News Release]; Aprea Therapeutics: Boston, MA, USA; Stockholm, Sweden, 2019; Available online: https://bit.ly/2IsKSKa (accessed on 16 April 2019).
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, J.F.; Sun, J.; Chen, L.; Yang, X.M.; Tang, H.Y.; Jing, Y.Y.; Kang, X.; He, Z.M.; Wu, J.Y.; et al. Histone H3K27 methylation modulates the dynamics of FANCD2 on chromatin to facilitate NHEJ and genome stability. J. Cell Sci. 2018, 131, jcs215525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Beggs, R.R.; Cooper, T.S.; Weaver, A.N.; Yang, E.S. Combining Chk1/2 Inhibition with Cetuximab and Radiation Enhances In Vitro and In Vivo Cytotoxicity in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2017, 16, 591–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werbrouck, C.; Evangelista, C.C.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 15, 6788–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagi, H.; Louis, N.; Unruh, D.; Sasaki, T.; He, X.; Zhang, A.; Ma, Q.; Piunti, A.; Shimazu, Y.; Lamano, J.B.; et al. Radiosensitization by Histone H3 Demethylase Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2019, 15, 5572–5583. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Chi, A.S.; Allen, J.E.; Oster, W.; Wen, P.Y.; Batchelor, T.T. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget 2017, 12, 79298–79304. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 13, 721–737. [Google Scholar] [CrossRef]
- Hall, M.D.; Odia, Y.; Allen, J.E.; Tarapore, R.; Khatib, Z.; Niazi, T.N.; Daghistani, D.; Schalop, L.; Chi, A.S.; Oster, W.; et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: A case report. J. Neurosurg. Pediatr. 2019, 5, 1–7. [Google Scholar] [CrossRef]
- Gardner, S.L.; Allen, J.C.; Zaky, W.T.; Odia, Y.; Daghistani, D.; Khatib, Z.; Koschmann, C.J.; Aguilera, D.; MacDonald, T.J.; Chi, A.S.; et al. ONC201 in previously-irradiated pediatric H3 K27M-mutant glioma. JCO 2019, 2019, 10046. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaev, A.; Fiveash, J.B.; Yang, E.S. Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG. Int. J. Mol. Sci. 2020, 21, 490. https://doi.org/10.3390/ijms21020490
Nikolaev A, Fiveash JB, Yang ES. Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG. International Journal of Molecular Sciences. 2020; 21(2):490. https://doi.org/10.3390/ijms21020490
Chicago/Turabian StyleNikolaev, Anatoly, John B. Fiveash, and Eddy S. Yang. 2020. "Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG" International Journal of Molecular Sciences 21, no. 2: 490. https://doi.org/10.3390/ijms21020490
APA StyleNikolaev, A., Fiveash, J. B., & Yang, E. S. (2020). Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG. International Journal of Molecular Sciences, 21(2), 490. https://doi.org/10.3390/ijms21020490