Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
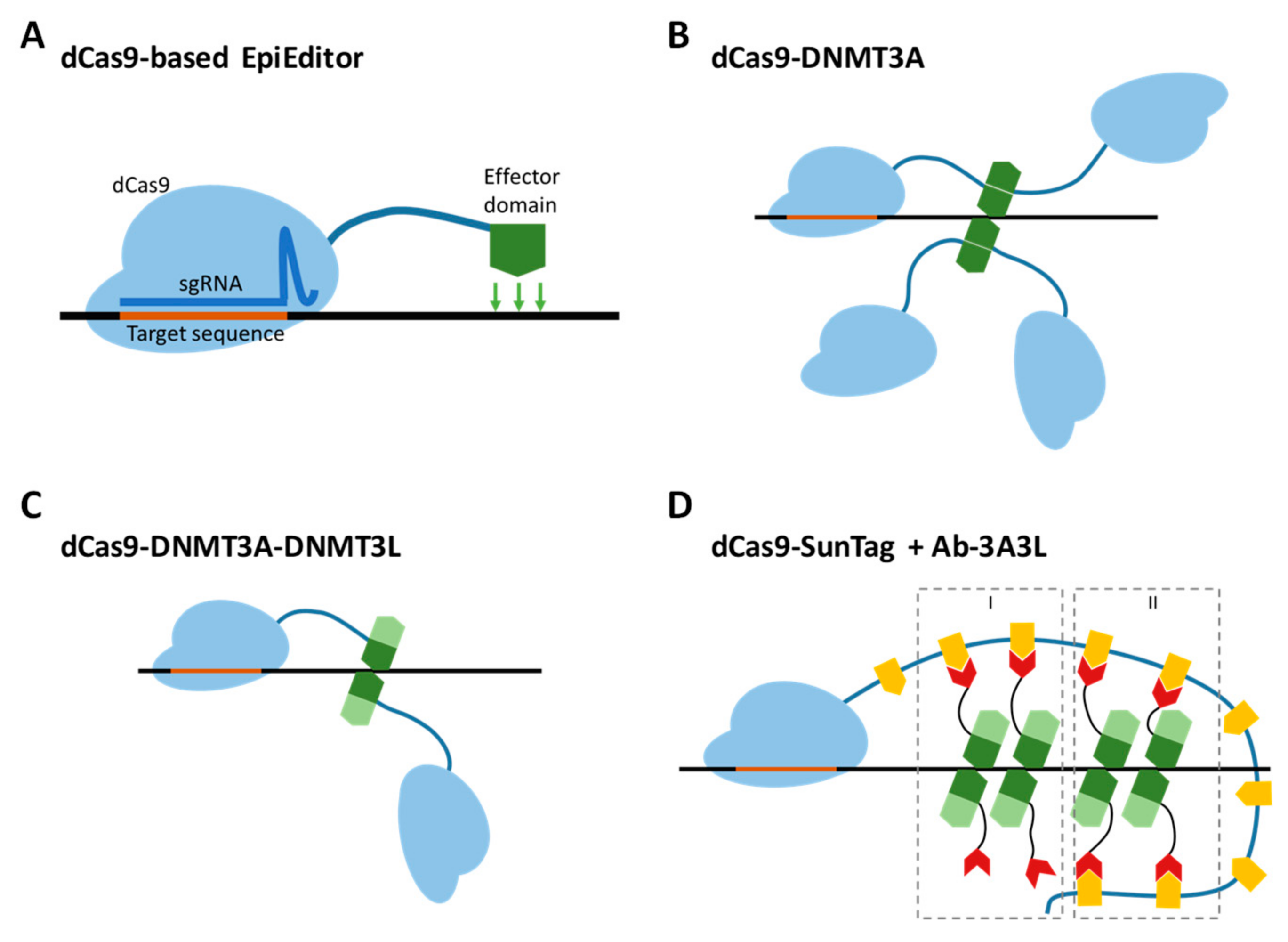
2.1. Comparison of the Efficiency and Specificity of the dCas9-DNMT3A-DNMT3L Direct Fusion with the dCas9-SunTag System
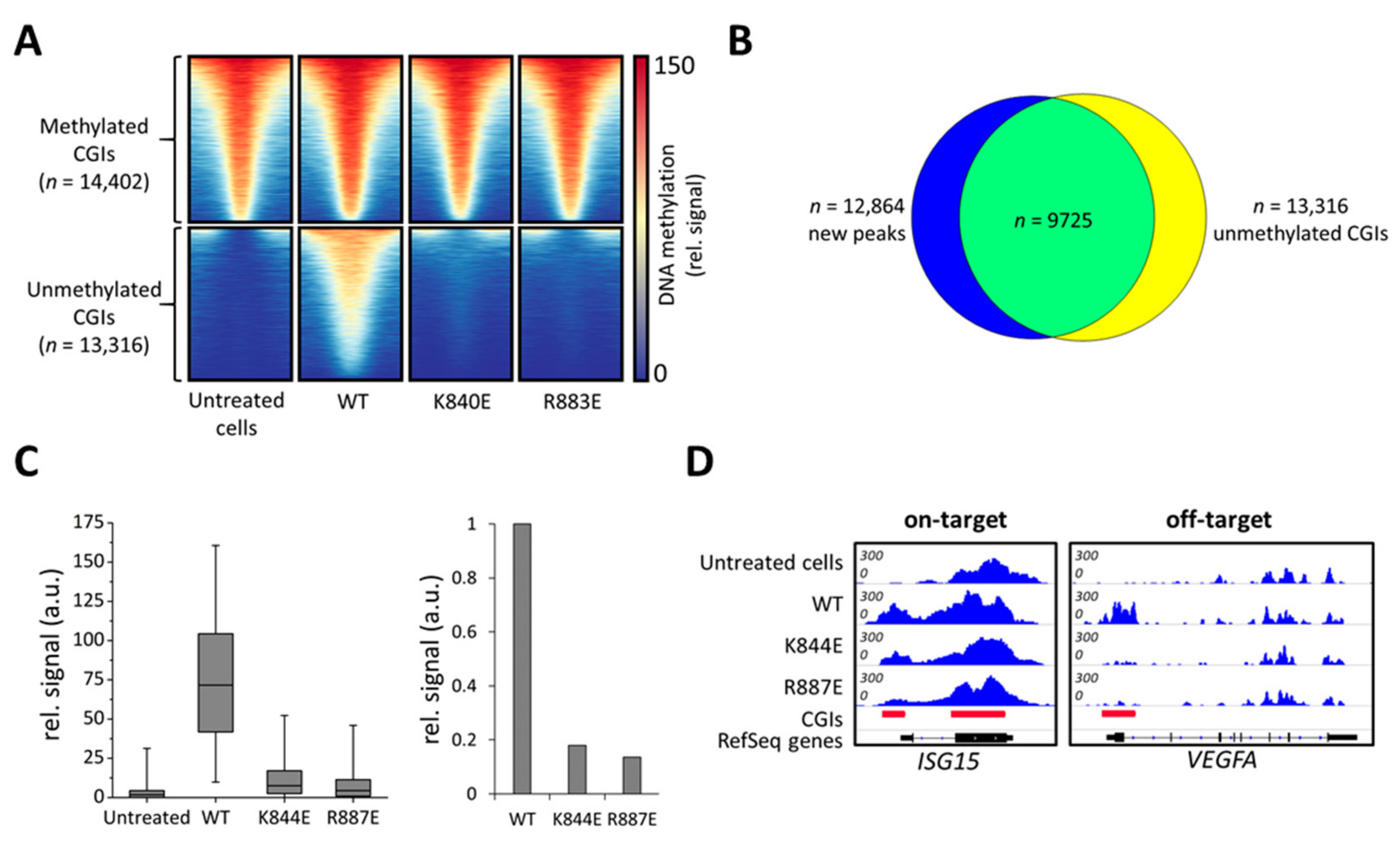
2.2. Rationally Designed Mutations in DNMT3A Decrease Off-Target Methylation
2.3. Additional Off-Target Methylation Experiments
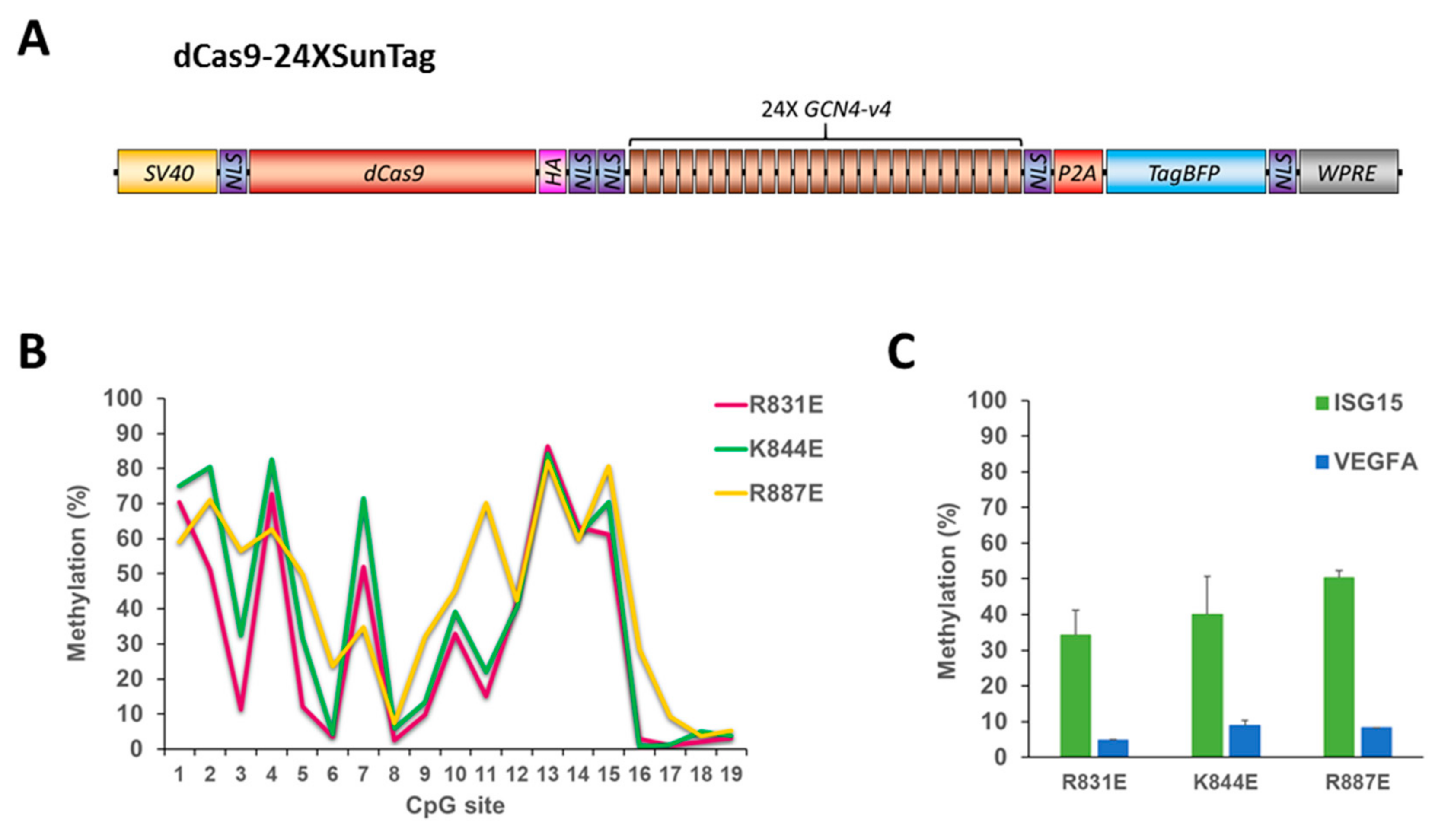
2.4. 24XSunTag Does Not Increase On-Target DNA Methylation
3. Discussion
4. Materials and Methods
4.1. Cloning
4.2. Site-Directed Mutagenesis
4.3. Cell Culture Co-Transfections and FACS
4.4. Targeted DNA Methylation Analysis
4.5. Genome-Wide DNA Methylation Analysis by MBD2-Pulldown Coupled with NGS
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mocellin, S.; Provenzano, M. RNA interference: Learning gene knock-down from cell physiology. J. Transl. Med. 2004, 2, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housden, B.E.; Muhar, M.; Gemberling, M.; Gersbach, C.A.; Stainier, D.Y.; Seydoux, G.; Mohr, S.E.; Zuber, J.; Perrimon, N. Loss-of-function genetic tools for animal models: Cross-species and cross-platform differences. Nat. Rev. Genet. 2017, 18, 24–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, Y.-H.H.; Goodell, M.A. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 2018, 19, 187. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Zhou, Y.; Sun, Q.; Tang, B. Novel Epigenetic Techniques Provided by the CRISPR/Cas9 System. Stem Cells Int. 2018, 2018, 7834175. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Briner, A.E.; Donohoue, P.D.; Gomaa, A.A.; Selle, K.; Slorach, E.M.; Nye, C.H.; Haurwitz, R.E.; Beisel, C.L.; May, A.P.; Barrangou, R. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol. Cell 2014, 56, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Papworth, M.; Minczuk, M.; Rohde, C.; Zhang, Y.; Ragozin, S.; Jeltsch, A. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 2007, 35, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunna, S.; Reinhardt, R.; Ragozin, S.; Jeltsch, A. Targeted Methylation of the Epithelial Cell Adhesion Molecule (EpCAM) Promoter to Silence Its Expression in Ovarian Cancer Cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kungulovski, G.; Nunna, S.; Thomas, M.; Zanger, U.M.; Reinhardt, R.; Jeltsch, A. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics Chromatin 2015, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolzenburg, S.; Beltran, A.S.; Swift-Scanlan, T.; Rivenbark, A.G.; Rashwan, R.; Blancafort, P. Stable oncogenic silencing in vivo by programmable and targeted de novo DNA methylation in breast cancer. Oncogene 2015, 34, 5427–5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef] [Green Version]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.; Nunna, S.; Rajavelu, A.; Zhang, Y.; Jurkowska, R.Z.; Reinhardt, R.; Rots, M.G.; Ragozin, S.; Jurkowski, T.P.; Jeltsch, A. Targeted Methylation and Gene Silencing of VEGF-A in Human Cells by Using a Designed Dnmt3a–Dnmt3L Single-Chain Fusion Protein with Increased DNA Methylation Activity. J. Mol. Biol. 2013, 425, 479–491. [Google Scholar] [CrossRef]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J. Clin. Investig. 2015, 125, 1998–2006. [Google Scholar] [CrossRef]
- Engmann, O.; Labonté, B.; Mitchell, A.; Bashtrykov, P.; Calipari, E.S.; Rosenbluh, C.; Loh, Y.-H.E.; Walker, D.M.; Burek, D.; Hamilton, P.J.; et al. Cocaine-Induced Chromatin Modifications Associate With Increased Expression and Three-Dimensional Looping of Auts2. Biol. Psychiatry 2017, 82, 794–805. [Google Scholar] [CrossRef]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galonska, C.; Charlton, J.; Mattei, A.L.; Donaghey, J.; Clement, K.; Gu, H.; Mohammad, A.W.; Stamenova, E.K.; Cacchiarelli, D.; Klages, S.; et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 2018, 9, 597. [Google Scholar] [CrossRef] [Green Version]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Liu, Y.; Xu, F.; Huang, J.; Daugaard, T.F.; Petersen, T.S.; Hansen, B.; Ye, L.; Zhou, Q.; Fang, F.; et al. Genome-wide determination of on-target and off-target characteristics for RNA-guided DNA methylation by dCas9 methyltransferases. GigaScience 2018, 7, 1–19. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.; Zhao, J.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, 201716945. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkowska, R.Z.; Anspach, N.; Urbanke, C.; Jia, D.; Reinhardt, R.; Nellen, W.; Cheng, X.; Jeltsch, A. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008, 36, 6656–6663. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.-S.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-M.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef]
- Gowher, H.; Loutchanwoot, P.; Vorobjeva, O.; Handa, V.; Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J. Mol. Biol. 2006, 357, 928–941. [Google Scholar] [CrossRef]
- Rajavelu, A.; Jurkowska, R.Z.; Fritz, J.; Jeltsch, A. Function and disruption of DNA methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res. 2012, 40, 569–580. [Google Scholar] [CrossRef]
- Lawhorn, I.E.; Ferreira, J.P.; Wang, C.L. Evaluation of sgRNA target sites for CRISPR-mediated repression of TP53. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Jeltsch, A.; Broche, J.; Lungu, C.; Bashtrykov, P. Biotechnological Applications of MBD Domain Proteins for DNA Methylation Analysis. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Xiong, T.; Rohm, D.; Workman, R.E.; Roundtree, L.; Novina, C.D.; Timp, W.; Ostermeier, M. Protein engineering strategies for improving the selective methylation of target CpG sites by a dCas9-directed cytosine methyltransferase in bacteria. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, A.; Lanio, T. Site-directed mutagenesis by polymerase chain reaction. Methods Mol. Biol. 2002, 182, 85–94. [Google Scholar] [CrossRef]
- Bashtrykov, P.; Jeltsch, A. DNA Methylation Analysis by Bisulfite Conversion Coupled to Double Multiplexed Amplicon-Based Next-Generation Sequencing (NGS). Methods Mol. Biol. 2018, 1767, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofacker, D.; Broche, J.; Laistner, L.; Adam, S.; Bashtrykov, P.; Jeltsch, A. Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects. Int. J. Mol. Sci. 2020, 21, 502. https://doi.org/10.3390/ijms21020502
Hofacker D, Broche J, Laistner L, Adam S, Bashtrykov P, Jeltsch A. Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects. International Journal of Molecular Sciences. 2020; 21(2):502. https://doi.org/10.3390/ijms21020502
Chicago/Turabian StyleHofacker, Daniel, Julian Broche, Laura Laistner, Sabrina Adam, Pavel Bashtrykov, and Albert Jeltsch. 2020. "Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects" International Journal of Molecular Sciences 21, no. 2: 502. https://doi.org/10.3390/ijms21020502
APA StyleHofacker, D., Broche, J., Laistner, L., Adam, S., Bashtrykov, P., & Jeltsch, A. (2020). Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects. International Journal of Molecular Sciences, 21(2), 502. https://doi.org/10.3390/ijms21020502