Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity



 , ,
, ,
Abstract
:1. Introduction
2. Results
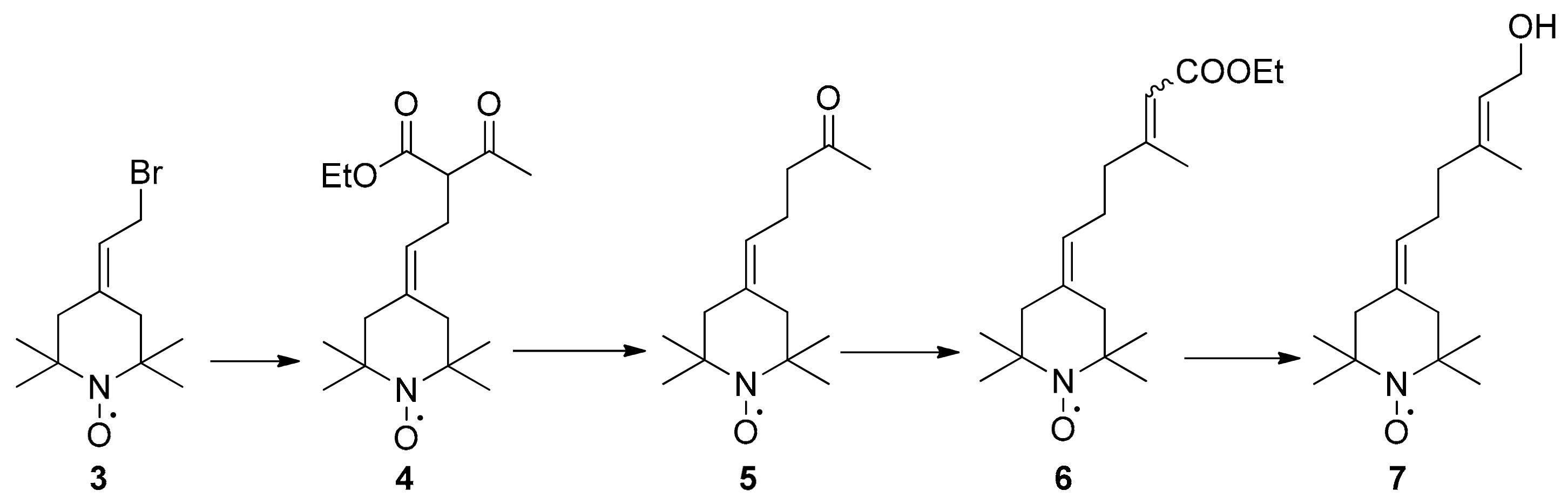
2.1. Synthesis of Spin-Labelled Geraniol (7)
2.2. Synthesis of Spin-Labelled Bergamottin (10)
2.3. Inhibition of the CYP Enzymes by BM and SL-BM
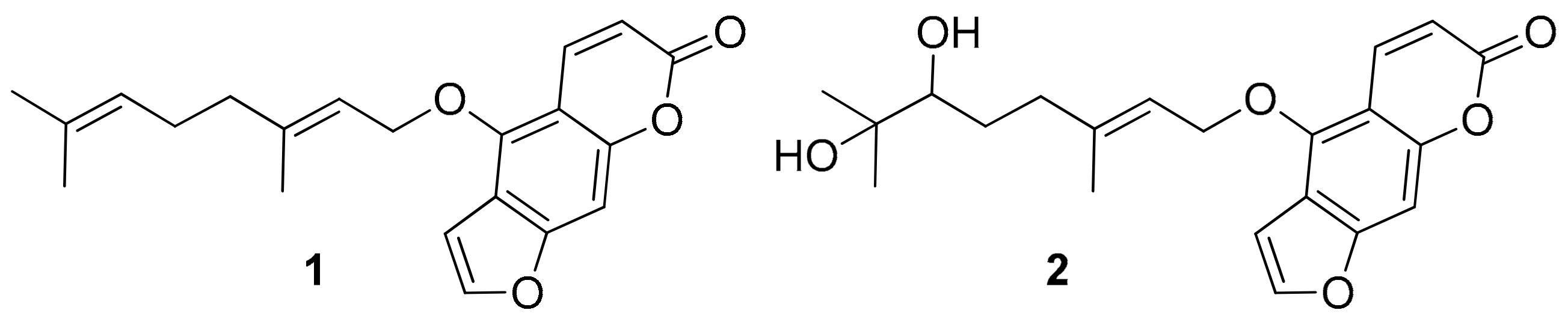
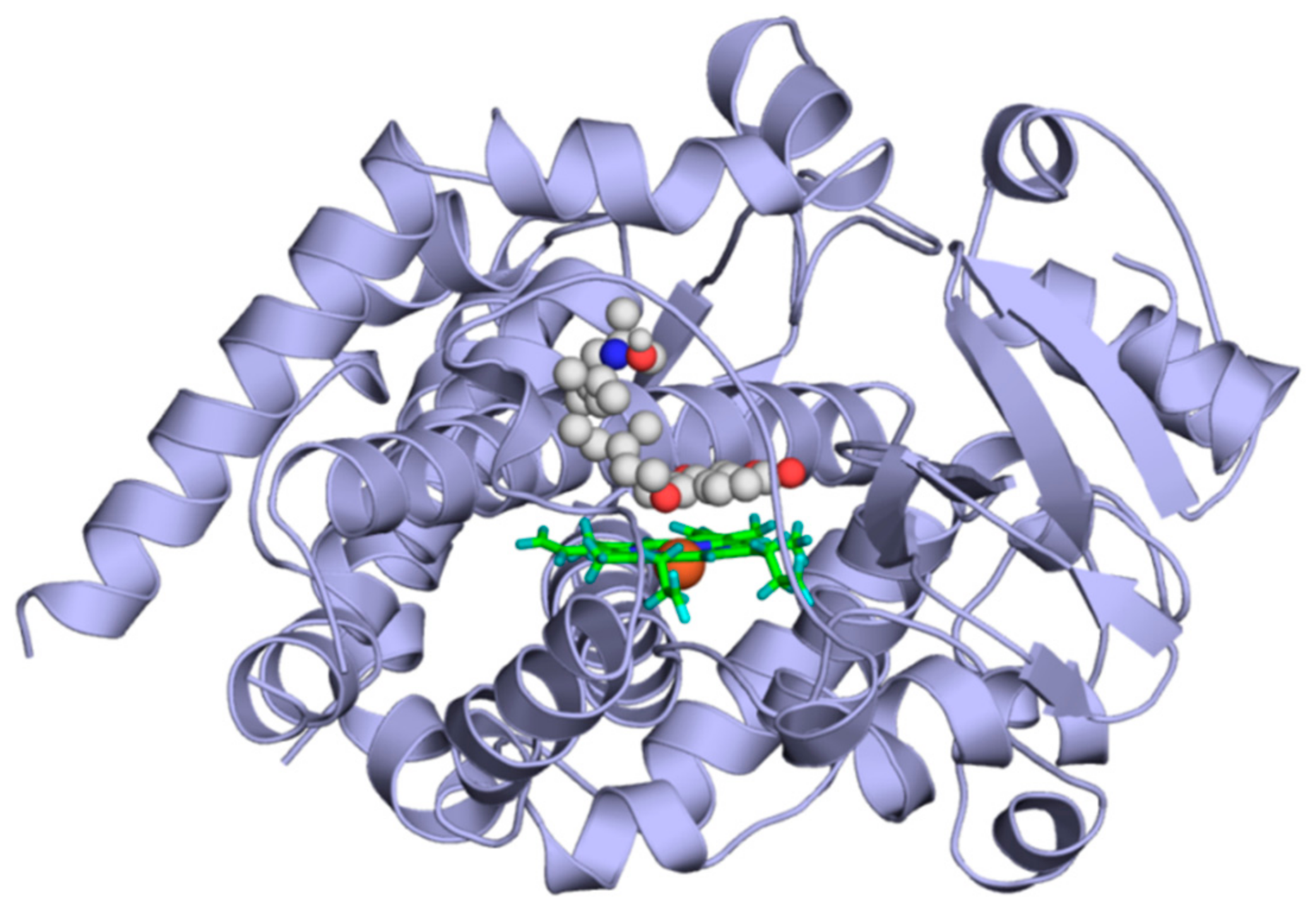
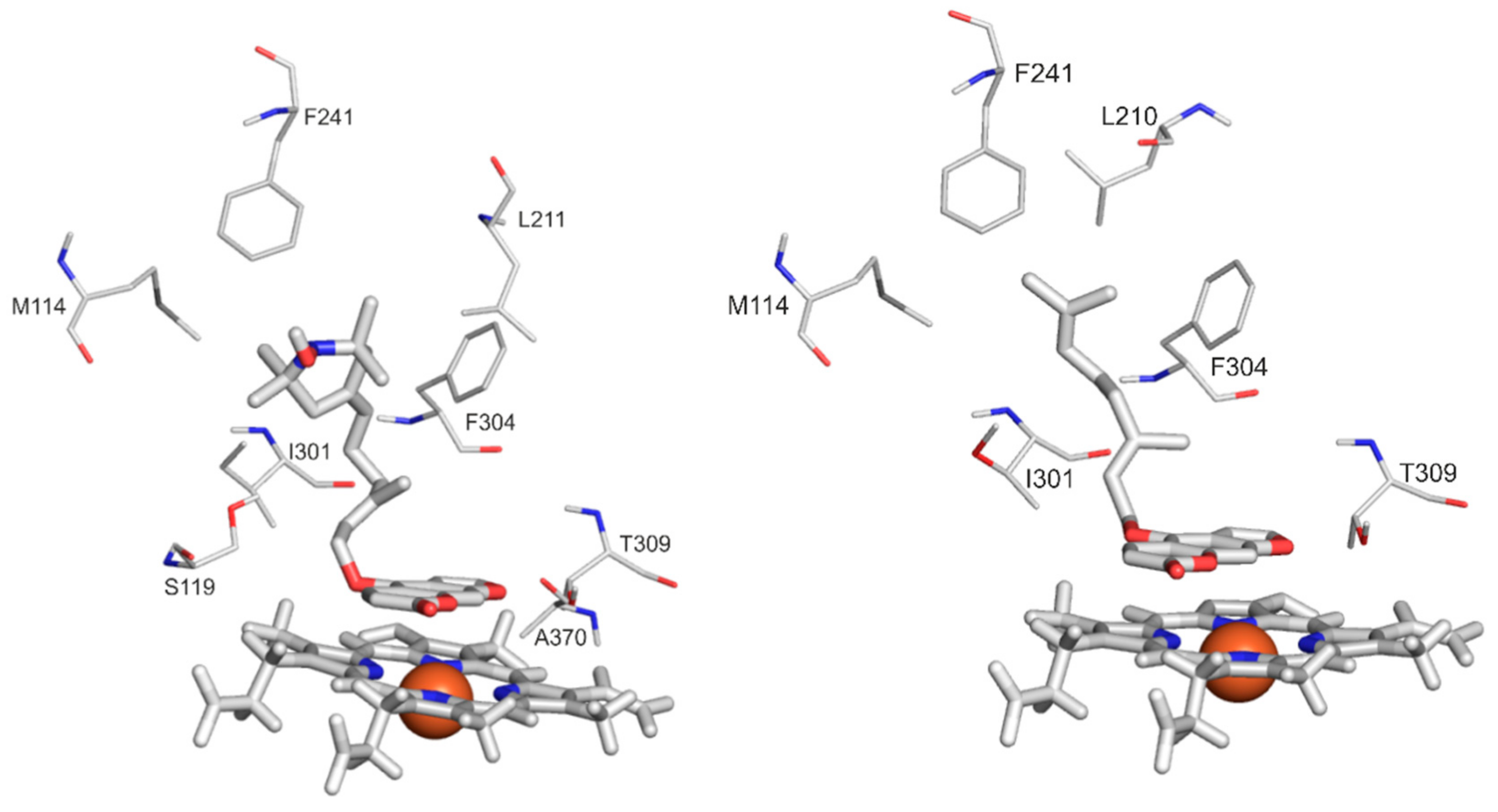
2.4. Modeling Studies
2.5. Cytotoxicity of BM and SL-BM
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General
4.1.2. Preparation of Compounds
4.2. CYP Inhibition Assays
4.2.1. Reagents
4.2.2. CYP Assays
4.3. Modeling Studies
4.3.1. Ligand Preparation
4.3.2. Target Preparation
4.3.3. Docking
4.4. Cell Viability Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Späth, E.; Kainrath, P. Über bergamottin und über die auffindung von limettin im bergamottöl (XXXIV. mitteil. über natürliche cumarine). Eur. J. Inorg. Chem. 1937, 70, 2272–2276. [Google Scholar] [CrossRef]
- Bailey, D.G.; Spence, J.D.; Munoz, C.; Arnold, J.M.O. Interaction of citrus juices with felodipine and nifedipine. Lancet 1991, 337, 268–269. [Google Scholar] [CrossRef]
- Weber, A.; Jager, R.; Borner, A.; Klinger, G.; Vollanth, R.; Mathey, K.; Balogh, A. Can grapefruit juice influence ethinylestradiol bioavailability. Contraception 1996, 53, 41–47. [Google Scholar] [CrossRef]
- Kuferschmidt, H.H.; Ha, H.R.; Ziegler, W.H.; Meier, P.J.; Krähenbühl, S. Interaction between grapefruit juice and midazolam in humans. Clin. Pharmacol. Ther. 1995, 58, 20–28. [Google Scholar] [CrossRef]
- Ducharme, M.P.; Warbasse, L.H.; Edwards, D. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. J. Clin. Phamacol. Ther. 1995, 40, 485–491. [Google Scholar] [CrossRef]
- Kantola, T.; Kivisto, K.T.; Neuvonen, P.J. Grapefruit juice greatly increases serum concentrations of lovastatin and lovastatin acid. Clin. Phamacol. Ther. 1998, 63, 397–402. [Google Scholar] [CrossRef]
- Lown, K.S.; Bailey, D.G.; Fontana, R.J.; Janardan, S.K.; Adair, C.H.; Fortlage, L.A.; Brown, M.B.; Gao, W.; Watkins, P.B. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J. Clin. Invest. 1997, 99, 2545–2553. [Google Scholar] [CrossRef]
- Bailey, D.G.; Arnold, J.; Arnold, O.; Spence, J.D. Grapefruit juice-drug interactions. Br. J. Clin. Pharmacol. 1998, 46, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Schmiedlin-Ren, P.; Edwards, D.J.; Fitzsimmons, M.E.; He, K.; Lown, K.S.; Woster, P.M.; Rahman, A.; Thummel, K.E.; Fisher, J.M.; Hollenberg, P.F.; et al. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug. Metab. Dispos. 1997, 25, 1228–1233. [Google Scholar]
- Guo, L.Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug. Metab. Dispos. 2000, 28, 766–771. [Google Scholar] [PubMed]
- Row, E.C.; Brow, S.A.; Stachulski, A.V.; Lennard, M.S. Design, synthesis and evaluation of furanocoumarin monomers as inhibitors of CYP3A4. Org. Biomol. Chem. 2006, 4, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, A.; Matsuo, H.; Yamada, S.; Takanaga, H.; Morimoto, S.; Shoyama, Y.; Ohtani, H.; Sawada, Y. Effect of furanocoumarin derivatives in grapefruit juice on the uptake of vinblastine by Caco-2 cells and on the activity of cytochrome P450 3A4. Br. J. Pharmacol. 2000, 130, 1369–1377. [Google Scholar] [CrossRef] [Green Version]
- Olguín-Reyes, S.; Camacho-Carranza, R.; Hernández-Ojeda, S.; Elinos-Baez, M.; Espinosa-Aguirre, J.J. Bergamottin is a competitive inhibitor of CYP1A1 and is antimutagenic in the Ames test. Food Chem. Toxicol. 2012, 50, 3094–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Iyer, K.R.; Hayes, R.N.; Sinz, M.W.; Woolf, T.F.; Hollenberg, P.F. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem. Res. Toxicol. 1998, 11, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Jeong-Hyeon, K.; Frank, A.; Gautam, S.; Kwang, S.A. Pharmacological Utilization of Bergamottin, Derived from Grapefruits, in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2018, 19, 4048–4060. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lee, J.H.; Sethi, G.; Kim, C.; Baek, S.H.; Nam, D.; Chung, W.S.; Kim, S.H.; Shim, B.S.; Ahn, K.S. Bergamottin, a natural furanocoumarin obtained from grapefruit juice induces chemosensitization and apoptosis through the inhibition of STAT3 signaling pathway in tumor cells. Cancer Lett. 2014, 354, 153–163. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Yun, H.J.; Choi, J.H.; Kang, K.W.; Jeong, H.G. Suppression of phorbol-12-myristate-13-acetate-induced tumor cell invasion by bergamottin via the inhibition of protein kinase Cδ/p38 mitogen-activated protein kinase and JNK/nuclear factor-κB-dependent matrix metalloproteinase-9 expression. Mol. Nutr. Food Res. 2010, 54, 977–990. [Google Scholar] [CrossRef]
- Wu, H.J.; Wu, H.B.; Zhao, Y.Q.; Chen, L.J.; Zou, H.Z. Bergamottin isolated from Citrus bergamia exerts in vitro and in vivo antitumor activity in lung adenocarcinoma through the induction of apoptosis, cell cycle arrest, mitochondrial membrane potential loss and inhibition of cell migration and invasion. Oncol. Rep. 2016, 36, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lee, E.J.; Lee, J.H.; Yang, W.M.; Nam, D.; Lee, J.H.; Lee, S.G.; Um, J.Y.; Shim, B.S.; Ahn, K.S. Simvastatin in combination with bergamottin potentiates TNF-induced apoptosis through modulation of NF-kappaB signalling pathway in human chronic myelogenous leukaemia. Pharm. Boil. 2016, 54, 2050–2060. [Google Scholar] [CrossRef] [Green Version]
- Krishna, M.C.; DeGraff, W.; Hankovszky, H.O.; Sár, P.C.; Kálai, T.; Jeko, J.; Russo, A.; Mitchell, J.B.; Hideg, K. Studies of Structure−Activity Relationship of Nitroxide Free Radicals and Their Precursors as Modifiers Against Oxidative Damage. J. Med. Chem. 1998, 41, 3477–3492. [Google Scholar] [CrossRef]
- Monti, E.; Cova, D.; Guido, E.; Morelli, R.; Oliva, C. Protective effect of the nitroxide tempol against the cardiotoxicity of adriamycin. Free Radic. Biol. Med. 1996, 21, 463–470. [Google Scholar] [CrossRef]
- Howard, B.J.; Yatin, S.; Hensley, K.; Allen, K.L.; Kelly, J.P.; Carney, J.; Butterfield, D.A. Prevention of Hyperoxia-Induced Alterations in Synaptosomal Membrane-Associated Proteins by N-tert-Butyl-α-Phenylnitrone and 4-Hydroxy-2,2,6,6-Tetramethylpiperidin-1-oxyl (Tempol). J. Neurochem. 1996, 67, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Deres, P.; Halmosi, R.; Tóth, A.; Kovács, K.; Pálfi, A.; Habon, T.; Czopf, L.; Kálai, T.; Hideg, K.; Sümegi, B.; et al. Prevention of doxorubicin-induced acute cardiotoxicity by an experimental antioxidant compound. J. Cardiovasc. Pharmacol. 2005, 45, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Suy, S.; Mitchell, J.B.; Ehleiter, D.; Haimovitz-Friedman, A.; Kasid, U. Nitroxides Tempol and Tempo Induce Divergent Signal Transduction Pathways in MDA-MB 231 Breast Cancer Cells. J. Biol. Chem. 1998, 273, 17871–17878. [Google Scholar] [CrossRef] [Green Version]
- Suy, S.; Mitchell, J.B.; Samuni, A.; Mueller, S.; Kasid, U. Nitroxide tempo, a small molecule, induces apoptosis in prostate carcinoma cells and suppresses tumor growth in athymic mice. Cancer 2005, 103, 1302–1313. [Google Scholar] [CrossRef]
- Nagane, M.; Yamashita, T.; Vörös, P.; Kálai, T.; Hideg, K.; Bognár, B. Synthesis and evaluation of paramagnetic caffeic acid phenethyl ester (CAPE) analogs. Monatsh. Chem. 2019, 150, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Kálai, T.; Borza, E.; Antus, C.; Radnai, B.; Gulyás-Fekete, G.; Fehér, A.; Sümegi, B.; Hideg, K. Synthesis and study of new paramagnetic resveratrol analogues. Bioorg. Med. Chem. 2011, 19, 7311–7317. [Google Scholar] [CrossRef]
- Dayton, A.; Selvendiran, K.; Meduru, S.; Khan, M.; Kuppusamy, M.L.; Naidu, S.; Kálai, T.; Hideg, K.; Kuppusamy, P. Amelioration of Doxorubicin-Induced Cardiotoxicity by an Anticancer-Antioxidant Dual-Function Compound HO-3867. J. Pharm. Exp. Ther. 2011, 339, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Kálai, T.; Kuppusamy, M.L.; Balog, M.; Selvendiran, K.; Rivera, B.K.; Kuppusamy, P.; Hideg, K. Synthesis of N-Substituted 3,5-Bis(arylidene)-4-piperidones with High Antitumor and Antioxidant Activity. J. Med. Chem. 2011, 54, 5414–5421. [Google Scholar] [CrossRef] [Green Version]
- Ravi, Y.; Selvendiran, K.; Naidu, S.; Meduru, S.; Citro, L.A.; Bognár, B.; Khan, M.; Kálai, T.; Hideg, K.; Kuppusamy, P.; et al. Pulmonary Hypertension Secondary to Left-Heart Failure Involves Peroxynitrite-Induced Downregulation of PTEN in the Lung. Hypertension 2013, 61, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Bognár, B.; Kuppusamy, M.L.; Madan, E.; Kálai, T.; Balog, M.; Jekő, J.; Kuppusamy, P.; Hideg, K. Synthesis and Biological Evaluation of Curcumin-Nitroxide-Based Molecular Hybrids as Antioxidant and Anti-Proliferative Agents. Med. Chem. 2017, 13, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. MOPAC2009, 2009; Steward Computational Chemistry: Colorado Springs, CO, USA, 2008. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girennavar, B.; Poulose, S.M.; Jayaprakasha, G.K.; Bhat, N.G.; Patil, B.S. Furocoumarins from grapefruit juice and their effect on human CYP 3A4 and CYP 1B1 isoenzymes. Bioorg. Med. Chem. 2006, 14, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Guo, L.Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch. Biochem. Biophys. 2000, 378, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Ito, H.; Ohnishi, R.; Hatano, T. Inhibitory effects of polyphenols on human cytochrome P450 3A4 and 2C9 activity. Food Chem. Toxicol. 2010, 48, 429–435. [Google Scholar] [CrossRef]
- Girennavar, B.; Jayaprakasha, G.K.; Patil, B.S. Potent inhibition of human cytochrome P450 3A4, 2D6, and 2C9 isoenzymes by grapefruit juice and its furocoumarins. J. Food. Sci. 2007, 72, 417–421. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, C.; Cao, Y.; Wang, Y.; Duan, W.; Xie, L.; Sun, C.; Li, X. Characterization and Purification of Bergamottin from Citrus grandis (L.) Osbeck cv. Yongjiazaoxiangyou and Its Antiproliferative Activity and Effect on Glucose Consumption in HepG2 cells. Molecules 2017, 22, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Poór, M.; Boda, G.; Needs, P.W.; Kroon, P.A.; Lemli, B.; Bencsik, T. Interaction of quercetin and its metabolites with warfarin: Displacement of warfarin from serum albumin and inhibition of CYP2C9 enzyme. Biomed. Pharmacother. 2017, 88, 574–581. [Google Scholar] [CrossRef]
- Poór, M.; Boda, G.; Mohos, V.; Kuzma, M.; Bálint, M.; Hetényi, C.; Bencsik, T. Pharmacokinetic interaction of diosmetin and silibinin with other drugs: Inhibition of CYP2C9-mediated biotransformation and displacement from serum albumin. Biomed. Pharmacother. 2018, 102, 912–921. [Google Scholar] [CrossRef]
- Mohos, V.; Bencsik, T.; Boda, G.; Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Poór, M. Interactions of casticin, ipriflavone, and resveratrol with serum albumin and their inhibitory effects on CYP2C9 and CYP3A4 enzymes. Biomed. Pharmacother. 2018, 107, 777–784. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Mohos, V.; Bencsik, T.; Lemli, B.; Kunsági-Máté, S.; Poór, M. Interactions of 7,8-Dihydroxyflavone with Serum Albumin as well as with CYP2C9, CYP2C19, CYP3A4, and Xanthine Oxidase Biotransformation Enzymes. Biomolecules 2019, 9, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LLC. Schrödinger Release 2019-3: Maestro, Schrödinger; LLC: New York, NY, USA, 2019; Available online: https://www.schrodinger.com/maestro. (accessed on 13 January 2020).
- Kálai, T.; Szabó, Z.; Jekő, J.; Hideg, K. Synthesis of New Allylic Nitroxides via the Wadsworth-Emmons Reaction. Org. Prep. Proced. Int. 1996, 28, 443–452. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Qian, G.; Wang, W.; Zhang, Y. Molecular docking to understand the metabolic behavior of GNF-351 by CYP3A4 and its potential drug–drug interaction with ketoconazole. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 235–238. [Google Scholar] [CrossRef]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. 3rd Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP2C9 Assay | Substrate Concentration (μM) | IC50 (μM) 1 | IC50(rel) 2 | α3 |
|---|---|---|---|---|
| Warfarin (positive ctrl) | 15 | 29.4 | 1.96 | 1.00 |
| Bergamottin | 15 | 14.7 | 2.94 | 0.50 |
| SL-Bergamottin | 15 | >60.0 | >4.00 | - |
| CYP2C19 Assay | Substrate Concentration (μM) | IC50 (μM) 1 | IC50(rel) 2 | α3 |
| Ticlopidine (positive ctrl) | 5 | 7.67 | 1.53 | 1.00 |
| Bergamottin | 5 | 1.01 | 0.20 | 0.13 |
| SL-Bergamottin | 5 | 14.6 | 2.92 | 1.90 |
| CYP3A4 Assay | Substrate Concentration (μM) | IC50 (μM) 1 | IC50(rel) 2 | α3 |
| Ketoconazole (positive ctrl) | 5 | 0.24 | 0.05 | 1.00 |
| Bergamottin | 5 | 2.04 | 0.41 | 8.50 |
| SL-Bergamottin | 5 | 0.40 | 0.08 | 1.67 |
| Ligand | KET | SL-BM | BM | |
|---|---|---|---|---|
| Rank of the docking of the ligand | 2 | 1 | 1 | |
| O–Fe dist (Å) | 4.2 | 4 | ||
| N–Fe dist (Å) | 3.8 | |||
| Calculated binding free energies ∆Gbind(kcal/mol) [33] | −9.4 | −10.4 | −9.2 | |
| List of interacting amino acidresidues | M114 | X | X | |
| S119 | X | |||
| L210 | X | |||
| L211 | X | |||
| F241 | X | X | ||
| I301 | X | X | ||
| F304 | X | X | X | |
| A305 | X | |||
| T309 | X | X | ||
| A370 | X | X | ||
| R372 | X | |||
| G481 | X | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsidó, B.Z.; Balog, M.; Erős, N.; Poór, M.; Mohos, V.; Fliszár-Nyúl, E.; Hetényi, C.; Nagane, M.; Hideg, K.; Kálai, T.; et al. Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity. Int. J. Mol. Sci. 2020, 21, 508. https://doi.org/10.3390/ijms21020508
Zsidó BZ, Balog M, Erős N, Poór M, Mohos V, Fliszár-Nyúl E, Hetényi C, Nagane M, Hideg K, Kálai T, et al. Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity. International Journal of Molecular Sciences. 2020; 21(2):508. https://doi.org/10.3390/ijms21020508
Chicago/Turabian StyleZsidó, Balázs Zoltán, Mária Balog, Nikolett Erős, Miklós Poór, Violetta Mohos, Eszter Fliszár-Nyúl, Csaba Hetényi, Masaki Nagane, Kálmán Hideg, Tamás Kálai, and et al. 2020. "Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity" International Journal of Molecular Sciences 21, no. 2: 508. https://doi.org/10.3390/ijms21020508
APA StyleZsidó, B. Z., Balog, M., Erős, N., Poór, M., Mohos, V., Fliszár-Nyúl, E., Hetényi, C., Nagane, M., Hideg, K., Kálai, T., & Bognár, B. (2020). Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity. International Journal of Molecular Sciences, 21(2), 508. https://doi.org/10.3390/ijms21020508