Enhanced Efficacy of PEGylated Liposomal Cisplatin: In Vitro and In Vivo Evaluation

 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Preparation of Nanoparticles
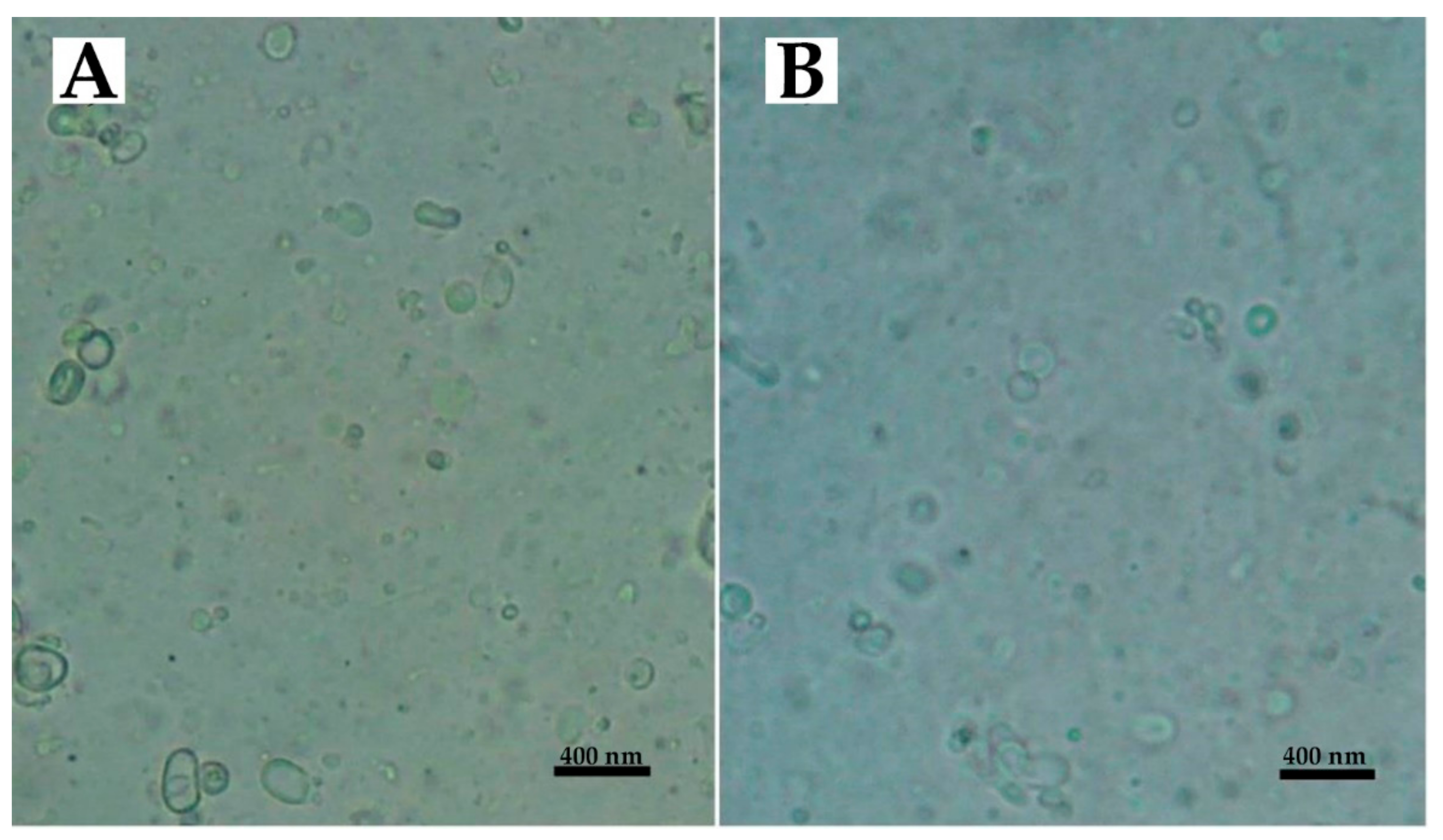
2.2. Characterization of Nanoparticles
2.3. Drug Release Study
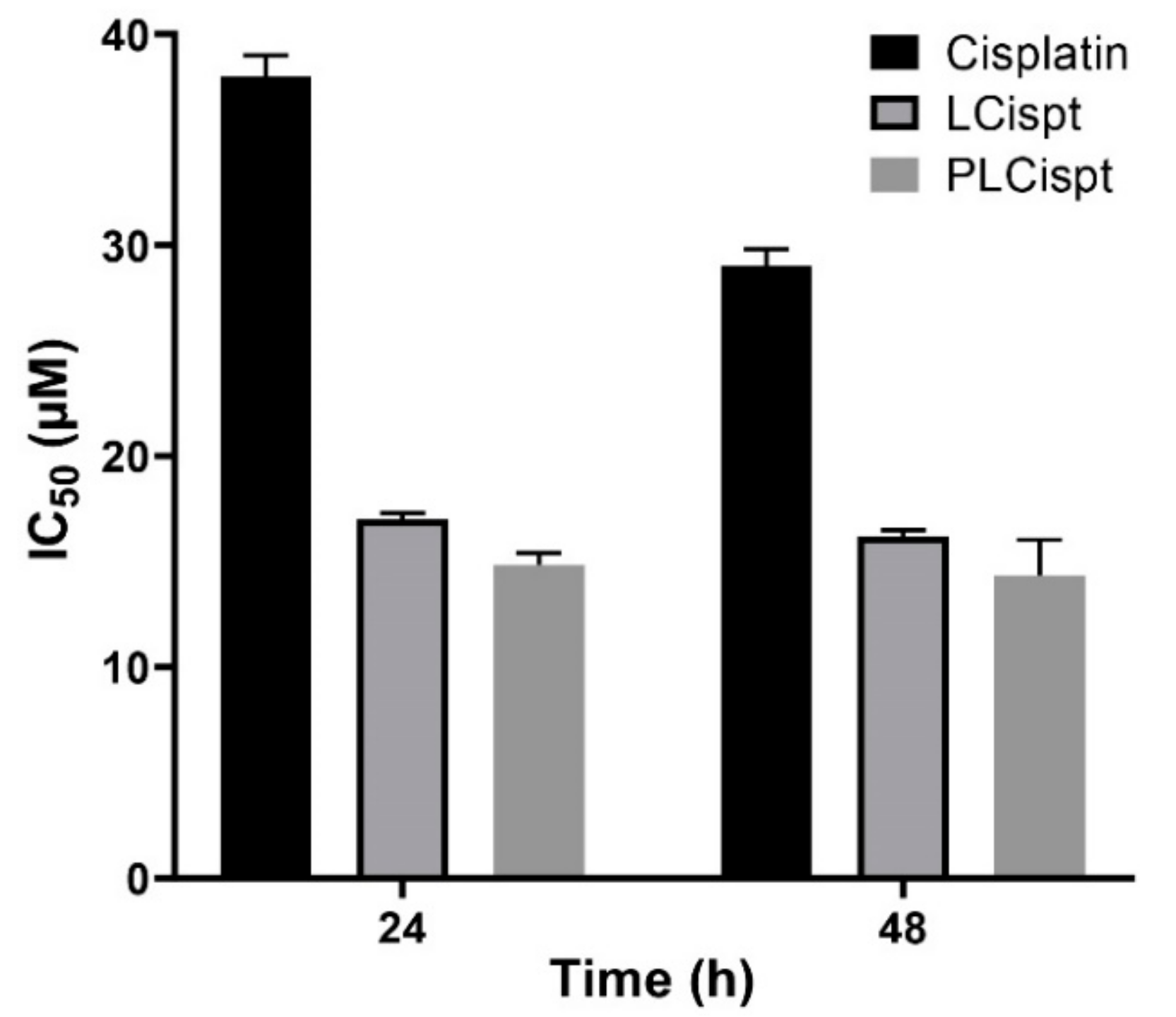
2.4. Cytotoxicity Study
2.5. Stability Study
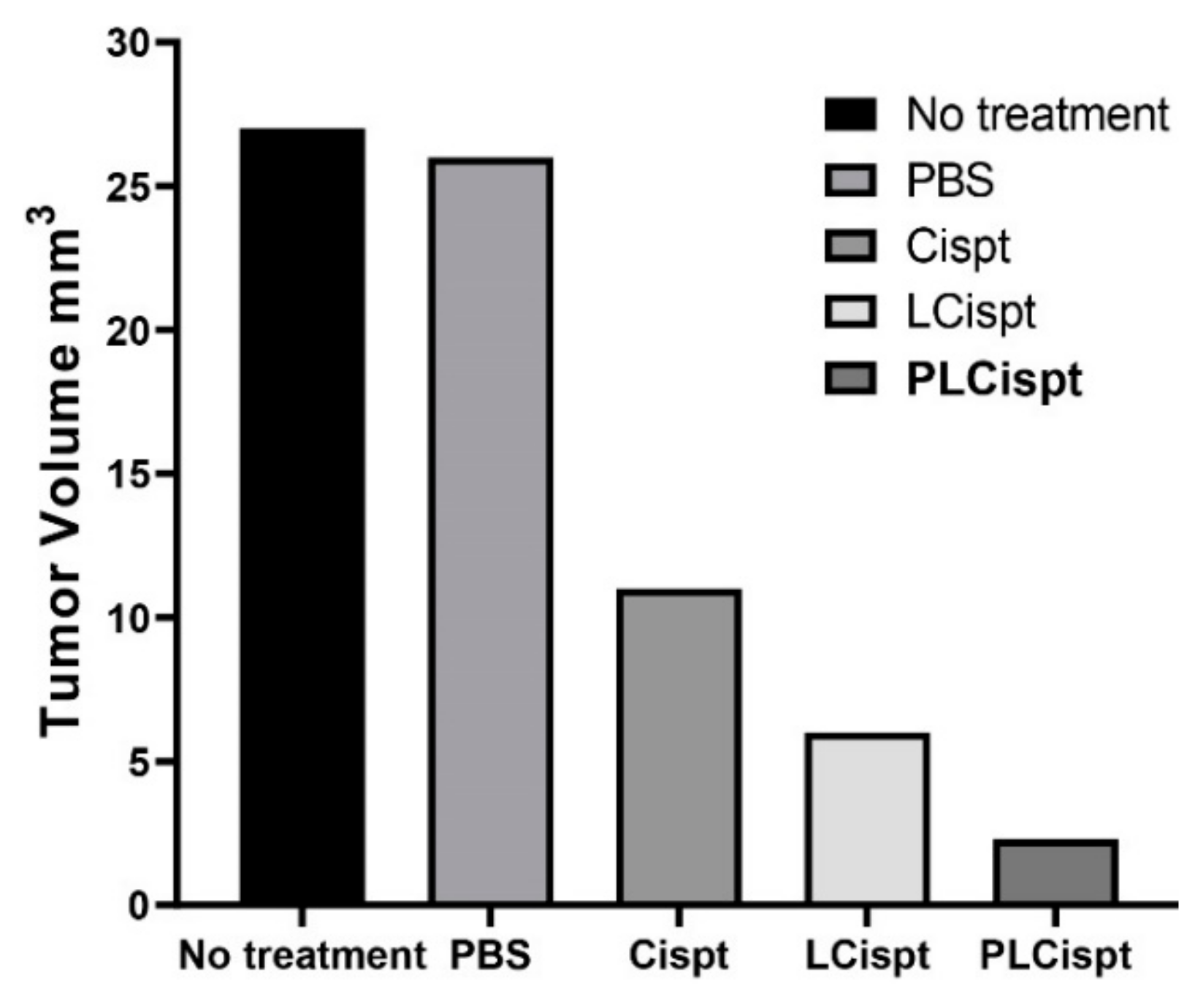
2.6. In Vivo Antitumor Efficacy of the Formulations
3. Materials and Methods
3.1. Materials
3.2. Preparation of Nanoparticles
3.3. Nanoparticles Characterization
3.4. Drug Release Study
3.5. Cytotoxicity Study
3.6. Stability Study
3.7. In Vivo Antitumor Efficacy of the Formulations
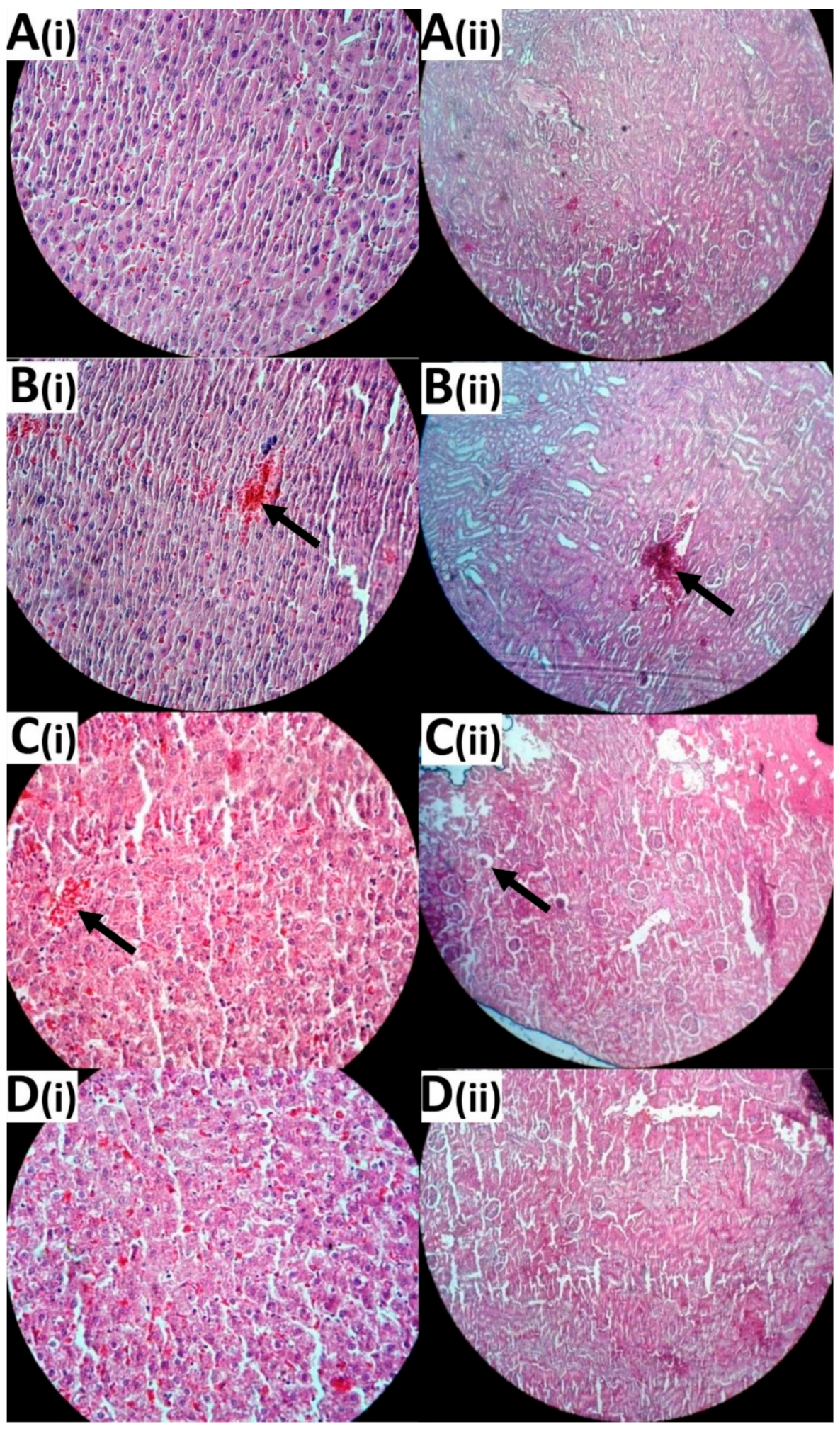
3.8. Histological Evaluation
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AAS | Atomic absorption spectroscopy |
| ATN | Acute tubular necrosis |
| BBN | N-butyl-N-(4-hydroxybutyl)-nitrosamine |
| Cispt | Cisplatin |
| FBS | Fetal bovine serum |
| H&E | Hematoxylin and eosin |
| IC50 | Half maximal inhibitory concentration |
| LCispt | Liposomal cisplatin |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate buffered saline |
| PCS | Photon correlation spectroscopy |
| PEG | Polyethylene glycol |
| PLCispt | PEGylated liposomal cisplatin |
| SEM | Scanning electron microscopy |
| TGII | Tumor growth inhibition index |
References
- Yu, E.Y.-W.; Wesselius, A.; van Osch, F.; Stern, M.C.; Jiang, X.; Kellen, E.; Lu, C.-M.; Pohlabeln, H.; Steineck, G.; Marshall, J. The association between coffee consumption and bladder cancer in the bladder cancer epidemiology and nutritional determinants (BLEND) international pooled study. Cancer Causes Control 2019, 30, 859–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Li, H.; Zou, D.; Gao, J. Ruguo key genes and tumor driving factors identification of bladder cancer based on the RNA-seq profile. OncoTargets Ther. 2016, 9, 2717. [Google Scholar]
- Oliveira, M.B.; Villa Nova, M.; Bruschi, M.L. A review of recent developments on micro/nanostructured pharmaceutical systems for intravesical therapy of the bladder cancer. Pharm. Dev. Technol. 2018, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guancial, E.A.; Rosenberg, J.E. The role of genomics in the management of advanced bladder cancer. Curr. Treat. Options Oncol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Ahmad, Z.; Majeed, S.; Shah, A. In vitro release and cytotoxicity of cisplatin loaded methoxy poly (ethylene glycol)-block-poly (glutamic acid) nanoparticles against human breast cancer cell lines. J. Drug Deliv. Sci. Technol. 2018, 43, 85–93. [Google Scholar] [CrossRef]
- Mandriota, G.; Di Corato, R.; Benedetti, M.; De Castro, F.; Fanizzi, F.P.; Rinaldi, R. Design and Application of Cisplatin-Loaded Magnetic Nanoparticle Clusters for Smart Chemotherapy. ACS Appl. Mater. Interfaces 2018, 11, 1864–1875. [Google Scholar] [CrossRef]
- Alavi, S.E.; Mansouri, H.; Koohi Moftakhari Esfahani, M.; Movahedi, F.; Akbarzadeh, A.; Chiani, M. Archaeosome: As new drug carrier for delivery of paclitaxel to breast cancer. Indian J. Clin. Biochem. 2014, 29, 150–153. [Google Scholar] [CrossRef] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Iinuma, H.; Maruyama, K.; Okinaga, K.; Sasaki, K.; Sekine, T.; Ishida, O.; Ogiwara, N.; Johkura, K.; Yonemura, Y. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int. J. Cancer 2002, 99, 130–137. [Google Scholar] [CrossRef]
- Kates, M.; Date, A.; Yoshida, T.; Afzal, U.; Kanvinde, P.; Babu, T.; Sopko, N.A.; Matsui, H.; Hahn, N.M.; McConkey, D.J. Preclinical Evaluation of Intravesical Cisplatin Nanoparticles for Non–Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2017, 23, 6592–6601. [Google Scholar] [CrossRef] [Green Version]
- Sudha, T.; Bharali, D.J.; Yalcin, M.; Darwish, N.H.; Coskun, M.D.; Keating, K.A.; Lin, H.-Y.; Davis, P.J.; Mousa, S.A. Targeted delivery of cisplatin to tumor xenografts via the nanoparticle component of nano-diamino-tetrac. Nanomedicine 2017, 12, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieler-Ferguson, H.M.; Chan, D.; Sockolosky, J.; Finney, L.; Maxey, E.; Vogt, S.; Szoka, F.C., Jr. Encapsulation, controlled release, and antitumor efficacy of cisplatin delivered in liposomes composed of sterol-modified phospholipids. Eur. J. Pharm. Sci. 2017, 103, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavizadeh, S.H.; Gheybi, F.; Nikpoor, A.R.; Badiee, A.; Golmohammadzadeh, S.; Jaafari, M.R. Therapeutic efficacy of cisplatin thermosensitive liposomes upon mild hyperthermia in C26 tumor bearing BALB/c mice. Mol. Pharm. 2017, 14, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, F.; Ebrahimi Shahmabadi, H.; Alavi, S.E.; Koohi Moftakhari Esfahani, M. Release modeling and comparison of nanoarchaeosomal, nanoliposomal and pegylated nanoliposomal carriers for paclitaxel. Tumor Biol. 2014, 35, 8665–8672. [Google Scholar] [CrossRef]
- Al Harthi, S.; Alavi, S.E.; Radwan, M.A.; El Khatib, M.M.; AlSarra, I.A. Nasal delivery of donepezil HCl-loaded hydrogels for the treatment of Alzheimer’s disease. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Gribko, A.; Künzel, J.; Wünsch, D.; Lu, Q.; Nagel, S.M.; Knauer, S.K.; Stauber, R.H.; Ding, G.-B. Is small smarter? Nanomaterial-based detection and elimination of circulating tumor cells: current knowledge and perspectives. Int. J. Nanomed. 2019, 14, 4187. [Google Scholar] [CrossRef] [Green Version]
- Zoghi, A.; Khosravi-Darani, K.; Omri, A. Process variables and design of experiments in liposome and nanoliposome research. Mini Rev. Med. Chem. 2018, 18, 324–344. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New developments in liposomal drug delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Mishra, P.; Nayak, B.; Dey, R. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 103–111. [Google Scholar] [CrossRef]
- Alavi, S.E.; Esfahani, M.K.M.; Ghassemi, S.; Akbarzadeh, A.; Hassanshahi, G. In vitro evaluation of the efficacy of liposomal and pegylated liposomal hydroxyurea. Indian J. Clin. Biochem. 2014, 29, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koohi Moftakhari Esfahani, M.; Alavi, S.E.; Movahedi, F.; Alavi, F.; Akbarzadeh, A. Cytotoxicity of liposomal Paclitaxel in breast cancer cell line mcf-7. Indian J. Clin. Biochem. 2013, 28, 358–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Su, X.; Yang, K.; Niu, F.; Li, J.; Song, J.; Chen, H.; Li, B.; Li, W.; Qian, W. CD20 antibody-conjugated immunoliposomes for targeted chemotherapy of melanoma cancer initiating cells. J. Biomed. Nanotechnol. 2015, 11, 1927–1946. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.; Liu, F.; He, X.; Ma, P.; Mao, L.; Gao, Y.; Yuan, F. The effect of sterol derivatives on properties of soybean and egg yolk lecithin liposomes: Stability, structure and membrane characteristics. Food Res. Int. 2018, 109, 24–34. [Google Scholar] [CrossRef]
- Bryła, A.; Lewandowicz, G.; Juzwa, W. Encapsulation of elderberry extract into phospholipid nanoparticles. J. Food Eng. 2015, 167, 189–195. [Google Scholar] [CrossRef]
- Alavi, S.E.; Cabot, P.J.; Moyle, P.M. Glucagon-like peptide-1 receptor agonists and strategies to improve their efficiency. Mol. Pharm. 2019, 16, 2278–2295. [Google Scholar] [CrossRef]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Huang, H.-L.; Li, J.; Qian, F.-C.; Li, L.-Q.; Niu, P.-P.; Dai, L.-C. Development of hybrid-type modified chitosan derivative nanoparticles for the intracellular delivery of midkine-siRNA in hepatocellular carcinoma cells. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 82–89. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, X.; Yang, B.; Deng, F.; Ji, J.; Yang, Y.; Huang, Z.; Zhang, X.; Wei, Y. Luminescence tunable fluorescent organic nanoparticles from polyethyleneimine and maltose: facile preparation and bioimaging applications. RSC Adv. 2014, 4, 22294–22298. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Miller, N. Phospholipid model membranes. I. Structural characteristics of hydrated liquid crystals. Biochim. Biophys. Acta BBA Biomembr. 1967, 135, 624–638. [Google Scholar] [CrossRef]
- Hatamihanza, H.; Alavi, S.E.; Ebrahimi Shahmabadi, H.; Akbarzadeh, A. Preparation, Characterization and Immunostimulatory Effects of CRD2 and CRD3 from TNF Receptor-1 Encapsulated into Pegylated Liposomal Nanoparticles. Int. J. Pept. Res. Ther. 2019, 1–9. [Google Scholar] [CrossRef]
- Koohi Moftakhari Esfahani, M.; Alavi, S.E.; Akbarzadeh, A.; Ghassemi, S.; Saffari, Z.; Farahnak, M.; Chiani, M. Pegylation of nanoliposomal paclitaxel enhances its efficacy in breast cancer. Trop. J. Pharm. Res. 2014, 13, 1195–1198. [Google Scholar] [CrossRef] [Green Version]
- Alavi, S.E.; Muflih Al Harthi, S.; Ebrahimi Shahmabadi, H.; Akbarzadeh, A. Cisplatin-Loaded Polybutylcyanoacrylate Nanoparticles with Improved Properties as an Anticancer Agent. Int. J. Mol. Sci. 2019, 20, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaei, F.; Alavi, S.E.; Ebrahimi Shahmabadi, H.; Akbarzadeh, A. Synthesis and characterization of polyethylene glycols conjugated to polybutylcyanoacrylate nanoparticles. Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 738–741. [Google Scholar] [CrossRef]
- Kouchakzadeh, H.; Shojaosadati, S.A.; Maghsoudi, A.; Farahani, E.V. Optimization of PEGylation conditions for BSA nanoparticles using response surface methodology. Aaps Pharmscitech 2010, 11, 1206–1211. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Chen, C.-M.; Lee, Y.-D. Synthesis of high loading and encapsulation efficient paclitaxel-loaded poly (n-butyl cyanoacrylate) nanoparticles via miniemulsion. Int. J. Pharm. 2007, 338, 267–275. [Google Scholar] [CrossRef]
- Duan, X.; He, C.; Kron, S.J.; Lin, W. Nanoparticle formulations of cisplatin for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 776–791. [Google Scholar] [CrossRef] [Green Version]
- McDaid, H.M.; Bhattacharya, S.K.; Chen, X.-T.; He, L.; Shen, H.-J.; Gutteridge, C.E.; Horwitz, S.B.; Danishefsky, S.J. Structure-activity profiles of eleutherobin analogs and their cross-resistance in Taxol-resistant cell lines. Cancer Chemother. Pharmacol. 1999, 44, 131–137. [Google Scholar] [CrossRef]
- Ebrahimi Shahmabadi, H.; Movahedi, F.; Koohi Moftakhari Esfahani, M.; Alavi, S.E.; Eslamifar, A.; Mohammadi Anaraki, G.; Akbarzadeh, A. Efficacy of Cisplatin-loaded polybutyl cyanoacrylate nanoparticles on the glioblastoma. Tumor Biol. 2014, 35, 4799–4806. [Google Scholar] [CrossRef]
- Munaweera, I.; Shi, Y.; Koneru, B.; Patel, A.; Dang, M.H.; Di Pasqua, A.J.; Balkus, K.J., Jr. Nitric oxide-and cisplatin-releasing silica nanoparticles for use against non-small cell lung cancer. J. Inorg. Biochem. 2015, 153, 23–31. [Google Scholar] [CrossRef]
- Porta-i-Batalla, M.; Eckstein, C.; Xifré-Pérez, E.; Formentín, P.; Ferré-Borrull, J.; Marsal, L.F. Sustained, controlled and stimuli-responsive drug release systems based on nanoporous anodic alumina with layer-by-layer polyelectrolyte. Nanoscale Res. Lett. 2016, 11, 372. [Google Scholar] [CrossRef] [Green Version]
- Fahr, A.; Liu, X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416. [Google Scholar] [CrossRef]
- Shen, S.I.; Jasti, B.R.; Li, X. Design of Controlled Release Drug Delivery Systems, 1st ed.; McGraw-Hill: New York, NY, USA, 2003; pp. 1–2. [Google Scholar]
- Kuang, Y.; Liu, J.; Liu, Z.; Zhuo, R. Cholesterol-based anionic long-circulating cisplatin liposomes with reduced renal toxicity. Biomaterials 2012, 33, 1596–1606. [Google Scholar] [CrossRef]
- Ou, H.; Cheng, T.; Zhang, Y.; Liu, J.; Ding, Y.; Zhen, J.; Shen, W.; Xu, Y.; Yang, W.; Niu, P. Surface-adaptive zwitterionic nanoparticles for prolonged blood circulation time and enhanced cellular uptake in tumor cells. Acta Biomater. 2018, 65, 339–348. [Google Scholar] [CrossRef]
- Liu, J.; Boonkaew, B.; Arora, J.; Mandava, S.H.; Maddox, M.M.; Chava, S.; Callaghan, C.; He, J.; Dash, S.; John, V.T. Comparison of Sorafenib-Loaded Poly (lactic/glycolic) Acid and DPPC Liposome Nanoparticles in the In Vitro Treatment of Renal Cell Carcinoma. J. Pharm. Sci. 2015, 104, 1187–1196. [Google Scholar] [CrossRef]
- Frangos, D.N.; Killion, J.J.; Fan, D.; Fishbeck, R.; Fidler, I.J. The development of liposomes containing interferon alpha for the intravesical therapy of human superficial bladder cancer. J. Urol. 1990, 143, 1252–1256. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, X.; Ashley, C.; Brinker, C.J. Electrostatically mediated liposome fusion and lipid exchange with a nanoparticle-supported bilayer for control of surface charge, drug containment, and delivery. J. Am. Chem. Soc. 2009, 131, 7567–7569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqahtani, M.S.; Alqahtani, A.; Thabit, A.; Syed, R. Novel lignin nanoparticles for oral drug delivery. J. Mater. Chem. B 2019, 7, 4461–4473. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, P.; Li, P.; Xue, A.; Zhang, X.; Zhang, H.; Jin, X. A magnetic chitosan hydrogel for sustained and prolonged delivery of Bacillus Calmette–Guérin in the treatment of bladder cancer. Biomaterials 2013, 34, 10258–10266. [Google Scholar] [CrossRef] [PubMed]
- Dornelas, C.A.; Fechine-Jamacaru, F.V.; Albuquerque, I.L.; Magalhães, H.I.F.; Souza, A.J.S.; Alves, L.A.; Almeida, P.R.C.; Lemos, T.L.G.; Castro, J.D.V.; Moraes, M.E.A. Chemoprevention with green propolis green propolis extracted in L-lysine versus carcinogenesis promotion with L-lysine in N-Butyl-N-[4-hydroxybutyl] nitrosamine (BBN) induced rat bladder cancer. Acta Cir. Bras. 2012, 27, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gofrit, O.N.; Birman, T.; Dinaburg, A.; Ayesh, S.; Ohana, P.; Hochberg, A. Chemically induced bladder cancer—A sonographic and morphologic description. Urology 2006, 68, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, J.; Nishiyama, H.; Yano, I.; Nakaya, A.; Kohama, H.; Kawai, K.; Joraku, A.; Nakamura, T.; Harashima, H.; Akaza, H. The therapeutic effects of R8-liposome-BCG-CWS on BBN-induced rat urinary bladder carcinoma. Anticancer Res. 2011, 31, 2065–2071. [Google Scholar] [PubMed]
- Dadashzadeh, S.; Mirahmadi, N.; Babaei, M.; Vali, A. Peritoneal retention of liposomes: Effects of lipid composition, PEG coating and liposome charge. J. Control. Release 2010, 148, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.; Fazlara, A.; Karamolah, M.H.; Shahriari, A.; Zadeh, H.N.; Pashmforosh, M. Effects of Echinacea purpurea on hepatic and renal toxicity induced by diethylnitrosamine in rats. Jundishapur J. Nat. Pharm. Prod. 2013, 8, 60. [Google Scholar] [CrossRef] [Green Version]
- Alavizadeh, S.H.; Akhtari, J.; Badiee, A.; Golmohammadzadeh, S.; Jaafari, M.R. Improved therapeutic activity of HER2 Affibody-targeted cisplatin liposomes in HER2-expressing breast tumor models. Expert Opin. Drug Deliv. 2016, 13, 325–336. [Google Scholar] [CrossRef]
- Potkul, R.K.; Gondal, J.; Bitterman, P.; Dretchen, K.L.; Rahman, A. Toxicities in rats with free versus liposomal encapsulated cisplatin. Am. J. Obstet. Gynecol. 1991, 164, 652–658. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batches of Nanoparticles | Size (nm) | Size Distribution | Zeta Potential (mV) | Lipid Composition |
|---|---|---|---|---|
| Liposome | 221.0 ± 11.0 | 0.40 ± 0.02 | −27.0 ± 1.3 | Lecithin and cholesterol |
| LCispt | 274.0 ± 12.6 | 0.058 ± 0.001 | −20.0 ± 0.9 | Lecithin and cholesterol |
| PLCispt | 251.0 ± 12.0 | 0.046 ± 0.002 | −7.0 ± 0.3 | Lecithin and cholesterol |
| Batches of Nanoparticles | Size (nm) | Size Distribution | Zeta Potential (mV) |
|---|---|---|---|
| PLCispt (Production day) | 251.0 ± 12.0 | 0.046 ± 0.002 | −7.0 ± 0.3 |
| PLCispt (Two months later) | 255.2 ± 13.0 | 0.049 ± 0.002 | −6.0 ± 0.3 |
| Group | Number of Animals | Organ | Score | BUN (mg/dL) | Creatinine (mg/dL) | ALP (U/L) | ALT (U/L) | AST (U/L) |
|---|---|---|---|---|---|---|---|---|
| Cispt | 10 | Liver | 2 | 67 ± 3.1 | 3.8 ± 0.2 | 273± 13.3 | 284 ± 14.3 | 365 ± 18.1 |
| Kidney | 1–2 | |||||||
| LCispt | 10 | Liver | 0–1 | 54 ± 2.6 | 3.1 ± 0.15 | 220± 11.1 | 237± 11.4 | 290± 14.3 |
| Kidney | 1 | |||||||
| PLCispt | 10 | Liver | 0–1 | 32 ± 1.5 | 1.8± 0.09 | 130± 6.2 | 138 ± 6.5 | 170± 8.2 |
| Kidney | 0–1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghaferi, M.; Asadollahzadeh, M.J.; Akbarzadeh, A.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Enhanced Efficacy of PEGylated Liposomal Cisplatin: In Vitro and In Vivo Evaluation. Int. J. Mol. Sci. 2020, 21, 559. https://doi.org/10.3390/ijms21020559
Ghaferi M, Asadollahzadeh MJ, Akbarzadeh A, Ebrahimi Shahmabadi H, Alavi SE. Enhanced Efficacy of PEGylated Liposomal Cisplatin: In Vitro and In Vivo Evaluation. International Journal of Molecular Sciences. 2020; 21(2):559. https://doi.org/10.3390/ijms21020559
Chicago/Turabian StyleGhaferi, Mohsen, Mohammad Javad Asadollahzadeh, Azim Akbarzadeh, Hasan Ebrahimi Shahmabadi, and Seyed Ebrahim Alavi. 2020. "Enhanced Efficacy of PEGylated Liposomal Cisplatin: In Vitro and In Vivo Evaluation" International Journal of Molecular Sciences 21, no. 2: 559. https://doi.org/10.3390/ijms21020559
APA StyleGhaferi, M., Asadollahzadeh, M. J., Akbarzadeh, A., Ebrahimi Shahmabadi, H., & Alavi, S. E. (2020). Enhanced Efficacy of PEGylated Liposomal Cisplatin: In Vitro and In Vivo Evaluation. International Journal of Molecular Sciences, 21(2), 559. https://doi.org/10.3390/ijms21020559