Pexophagy: A Model for Selective Autophagy

Abstract
:1. Introduction to Peroxisomes
2. Peroxisome Homeostasis
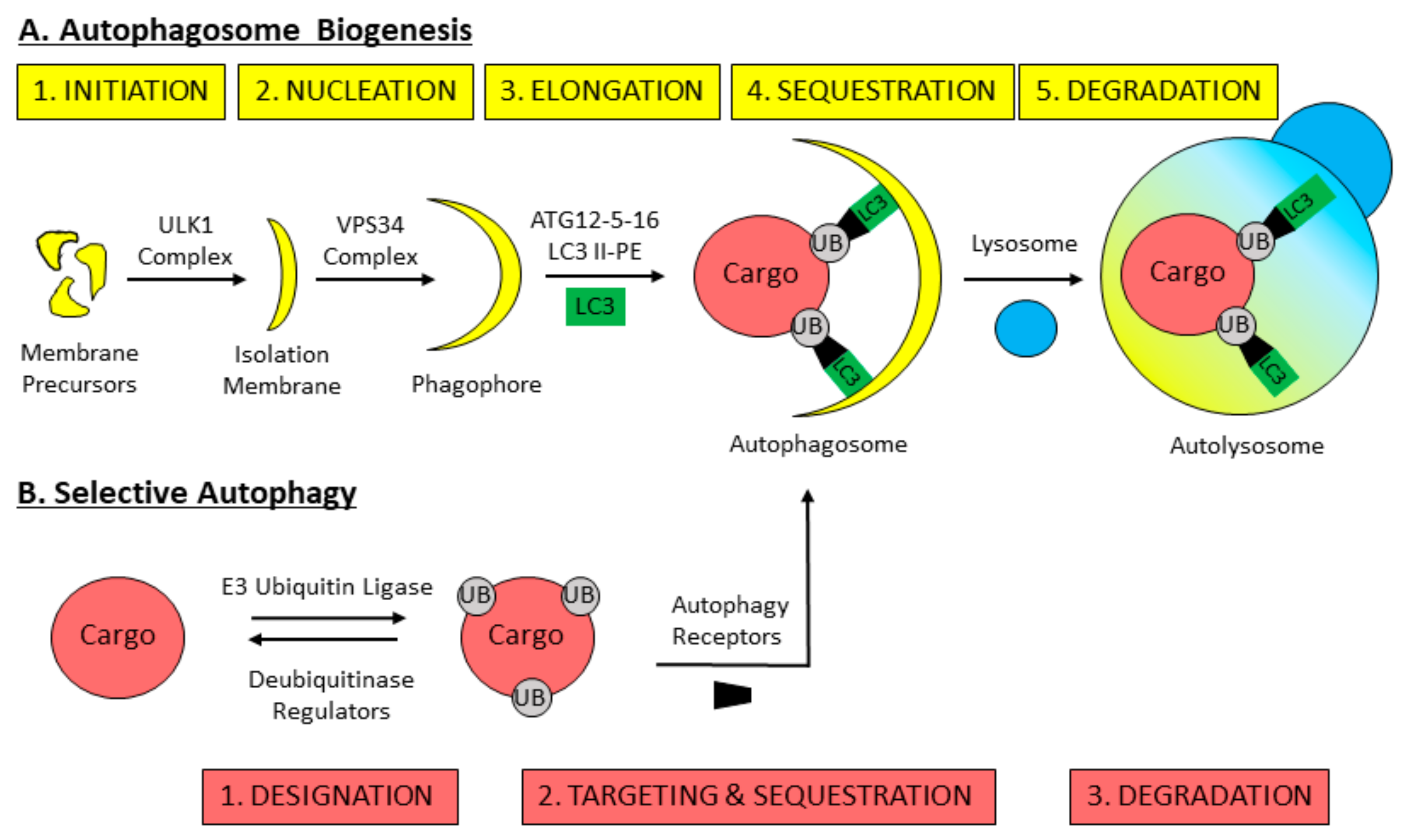
3. Mechanisms of Macroautophagy
4. Selective Autophagy Overview
5. Pexophagy: The Selective Autophagic Degradation of Peroxisomes
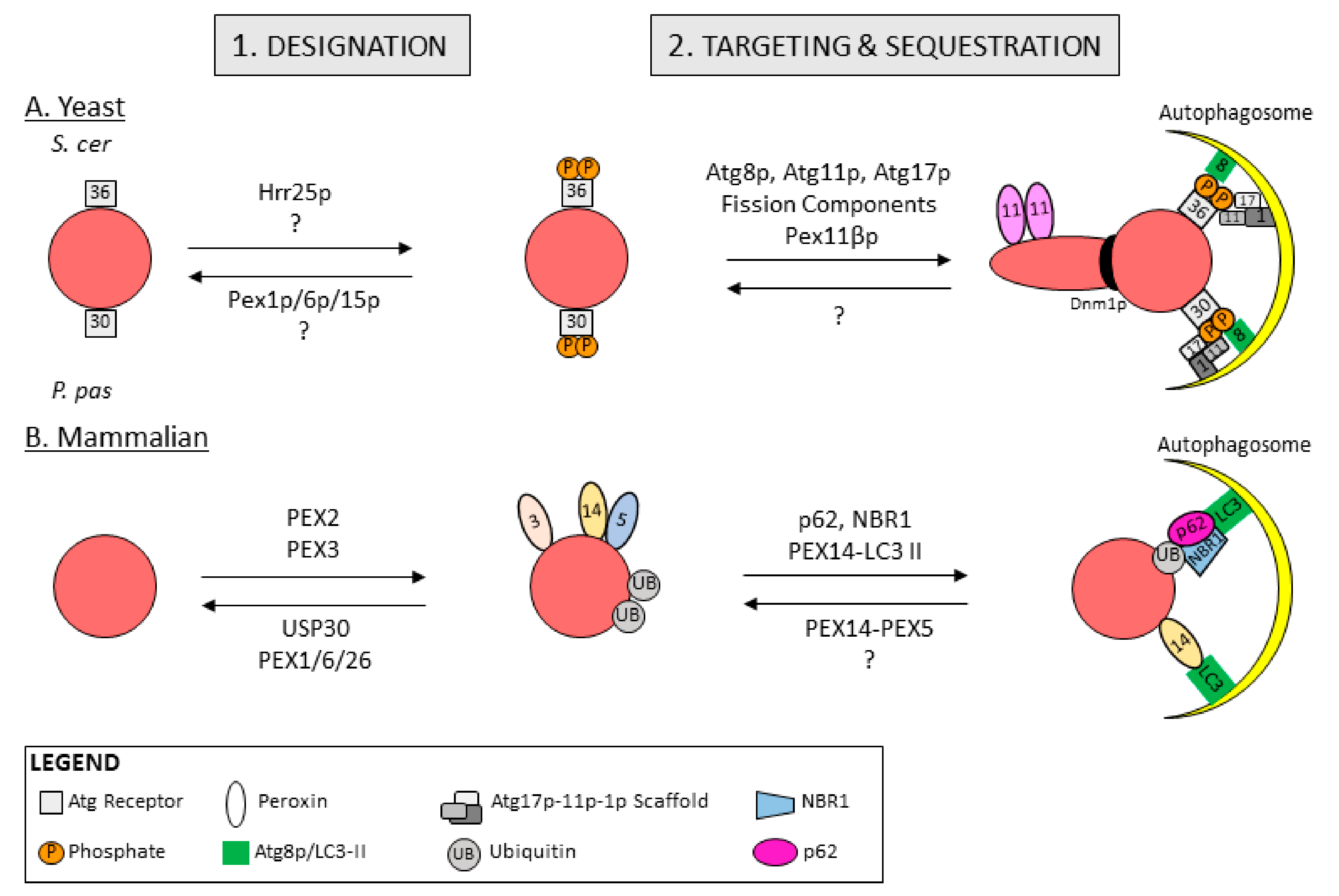
5.1. Peroxisome Designation for Degradation in Yeast
5.2. Peroxisome Designation for Degradation in Mammals
5.3. Peroxisome Targeting and Sequestration
5.4. Peroxisome Degradation
6. Regulation of Pexophagy
7. Pexophagy in Human Health and Disease
8. Perspectives
8.1. Yeast vs. Mammalian Pexophagy
8.2. Peroxisomes as a Model for Selective Autophagy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VLCFA | Very long chain fatty acid |
| ROS | Reactive oxygen species |
| ER | Endoplasmic Reticulum |
| PMP | Peroxisome membrane protein |
| PTS | Peroxisome targeting signal |
| CMA | Chaperone mediated autophagy |
| PAS | Phagophore assembly site |
| AIM | Atg8-interacting motif |
| LIR | LC3-interacting region |
| UIM | Ubiquitin-interacting motif |
| RPC | Receptor protein complex |
| UBA | Ubiquitin associated domain |
| CHO | Chinese hamster ovary |
| PIP | Phosphatidylinositol phosphate |
| PBD | Peroxisome Biogenesis Disorder |
| PD | Parkinson’s Disease |
References
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B.; De Duve, C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalik, S.J.; Rainville, A.M.; Watkins, P.A. Phytanic Acid α-oxidation in Rat Liver Peroxisomes: Production of α-hydroxyphytanoyl-CoA and Formate is Enhanced by Dioxygenase. Eur. J. Biochem. 1995, 232, 545–551. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, W.J.; Tape, K.N.; Shackelford, J.E.; Duan, X.; Kasumov, T.; Kelleher, J.K.; Brunengraber, H.; Krisans, S.K. Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem. Cell Biol. 2007, 127, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Knoblach, B.; Sun, X.; Coquelle, N.; Fagarasanu, A.; Poirier, R.L.; Rachubinski, R.A. An ER-peroxisome tether exerts peroxisome population control in yeast. EMBO J. 2013, 32, 2439–2453. [Google Scholar] [CrossRef] [Green Version]
- Hua, R.; Cheng, D.; Coyaud, É.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Klug, Y.A.; Dimitrov, L.; Erez, Z.; Chuartzman, S.G.; Elinger, D.; Yofe, I.; Soliman, K.; Gärtner, J.; Thoms, S.; et al. Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 2014, 10, 1742–1748. [Google Scholar] [CrossRef]
- Mattiazzi Ušaj, M.; Brložnik, M.; Kaferle, P.; Žitnik, M.; Wolinski, H.; Leitner, F.; Kohlwein, S.D.; Zupan, B.; Petrovič, U. Genome-Wide Localization Study of Yeast Pex11 Identifies Peroxisome-Mitochondria Interactions through the ERMES Complex. J. Mol. Biol. 2015, 427, 2072–2087. [Google Scholar] [CrossRef]
- Chu, B.-B.; Liao, Y.-C.; Qi, W.; Xie, C.; Du, X.; Wang, J.; Yang, H.; Miao, H.-H.; Li, B.-L.; Song, B.-L. Cholesterol Transport through Lysosome-Peroxisome Membrane Contacts. Cell 2015, 161, 291–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binns, D.; Januszewski, T.; Chen, Y.; Hill, J.; Markin, V.S.; Zhao, Y.; Gilpin, C.; Chapman, K.D.; Anderson, R.G.W.; Goodman, J.M. An intimate collaboration between peroxisomes and lipid bodies. J. Cell Biol. 2006, 173, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M. Tubulo-reticular clusters of peroxisomes in living COS-7 cells: Dynamic behavior and association with lipid droplets. J. Histochem. Cytochem. 2001, 49, 1421–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.O.; Trimble, R.; Guo, F.; Mak, H.Y. Lipid droplets as ubiquitous fat storage organelles in C. elegans. BMC Cell Biol. 2010, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [Green Version]
- Mast, F.D.; Aitchison, J.D. Characterization of Peroxisomal Regulation Networks. Subcell. Biochem. 2018, 89, 367–382. [Google Scholar]
- Akşit, A.; van der Klei, I.J. Yeast peroxisomes: How are they formed and how do they grow? Int. J. Biochem. Cell Biol. 2018, 105, 24–34. [Google Scholar] [CrossRef]
- Farré, J.-C.; Mahalingam, S.S.; Proietto, M.; Subramani, S. Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 2019, 20, e46864. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.K.; Mullen, R.T.; Schumann, U.; Lippincott-Schwartz, J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J. Cell Biol. 2006, 173, 521–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Morrell, J.C.; Jones, J.M.; Gould, S.J. PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins. J. Cell Biol. 2004, 164, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J. Cell Biol. 2004, 164, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titorenko, V.I.; Rachubinski, R.A. The life cycle of the peroxisome. Nat. Rev. Mol. Cell Biol. 2001, 2, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef]
- Koch, J.; Pranjic, K.; Huber, A.; Ellinger, A.; Hartig, A.; Kragler, F.; Brocard, C. PEX11 family members are membrane elongation factors that coordinate peroxisome proliferation and maintenance. J. Cell Sci. 2010, 123, 3389–3400. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M.; Reuber, B.E.; Morrell, J.C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T.A.; Valle, D.; Schroer, T.A.; Gould, S.J. Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli. J. Biol. Chem. 1998, 273, 29607–29614. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Takeshita, A.; Tsukamoto, T.; Gonzalez, F.J.; Osumi, T. Tissue-selective, bidirectional regulation of PEX11 alpha and perilipin genes through a common peroxisome proliferator response element. Mol. Cell. Biol. 2004, 24, 1313–1323. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.; Prinz, W.A. Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 15785–15790. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, Y.; Rachubinski, R.A.; Lazarow, P.B. Synthesis of a major integral membrane polypeptide of rat liver peroxisomes on free polysomes. Proc. Natl. Acad. Sci. USA 1984, 81, 7127–7131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, S.J.; Keller, G.-A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989, 108, 1657–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinkels, B.W.; Gould, S.J.; Bodnar, A.G.; Rachubinski, R.A.; Subramani, S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991, 10, 3255–3262. [Google Scholar] [CrossRef]
- Dammai, V.; Subramani, S. The human peroxisomal targeting signal receptor, Pex5p, is translocated into the peroxisomal matrix and recycled to the cytosol. Cell 2001, 105, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Braverman, N.; Steel, G.; Obie, C.; Moser, A.; Moser, H.; Gould, S.J.; Valle, D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 1997, 15, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.E.; Schliebs, W. Pex14p, more than just a docking protein. Biochim. Biophys. Acta 2006, 1763, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Distel, B. Pex13p: Docking or cargo handling protein? Biochim. Biophys. Acta 2006, 1763, 1585–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinecke, M.; Cizmowski, C.; Schliebs, W.; Krüger, V.; Beck, S.; Wagner, R.; Erdmann, R. The peroxisomal importomer constitutes a large and highly dynamic pore. Nat. Cell Biol. 2010, 12, 273–277. [Google Scholar] [CrossRef]
- Platta, H.W.; Grunau, S.; Rosenkranz, K.; Girzalsky, W.; Erdmann, R. Functional role of the AAA peroxins in dislocation of the cycling PTS1 receptor back to the cytosol. Nat. Cell Biol. 2005, 7, 817–822. [Google Scholar] [CrossRef]
- Platta, H.W.; Girzalsky, W.; Erdmann, R. Ubiquitination of the peroxisomal import receptor Pex5p. Biochem. J. 2004, 384, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sá-Miranda, C.; Azevedo, J.E. Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J. Biol. Chem. 2007, 282, 31267–31272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, J.A.K.W.; Emmrich, K.; Meyer, H.E.; Kunau, W.-H. Ubiquitination of the peroxisomal targeting signal type 1 receptor, Pex5p, suggests the presence of a quality control mechanism during peroxisomal matrix protein import. J. Biol. Chem. 2005, 280, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Magraoui, F.; Brinkmeier, R.; Mastalski, T.; Hupperich, A.; Strehl, C.; Schwerter, D.; Girzalsky, W.; Meyer, H.E.; Warscheid, B.; Erdmann, R.; et al. The deubiquitination of the PTS1-import receptor Pex5p is required for peroxisomal matrix protein import. Biochim. Biophys. Acta 2019, 1866, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Grou, C.P.; Francisco, T.; Rodrigues, T.A.; Freitas, M.O.; Pinto, M.P.; Carvalho, A.F.; Domingues, P.; Wood, S.A.; Rodríguez-Borges, J.E.; Sá-Miranda, C.; et al. Identification of ubiquitin-specific protease 9X (USP9X) as a deubiquitinase acting on ubiquitin-peroxin 5 (PEX5) thioester conjugate. J. Biol. Chem. 2012, 287, 12815–12827. [Google Scholar] [CrossRef] [Green Version]
- Tsilibaris, V.; Maenhaut-Michel, G.; Van Melderen, L. Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 2006, 157, 701–713. [Google Scholar] [CrossRef]
- Farmer, L.M.; Rinaldi, M.A.; Young, P.G.; Danan, C.H.; Burkhart, S.E.; Bartel, B. Disrupting autophagy restores peroxisome function to an Arabidopsis lon2 mutant and reveals a role for the LON2 protease in peroxisomal matrix protein degradation. Plant Cell 2013, 25, 4085–4100. [Google Scholar] [CrossRef] [Green Version]
- Bartel, B.; Farmer, L.M.; Rinaldi, M.A.; Young, P.G.; Danan, C.H.; Burkhart, S.E. Mutation of the Arabidopsis LON2 peroxisomal protease enhances pexophagy. Autophagy 2014, 10, 518–519. [Google Scholar] [CrossRef] [Green Version]
- Van Leyen, K.; Duvoisin, R.M.; Engelhardt, H.; Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 1998, 395, 392–395. [Google Scholar] [CrossRef]
- Yokota, S.; Dariush Fahimi, H. Degradation of excess peroxisomes in mammalian liver cells by autophagy and other mechanisms. Histochem. Cell Biol. 2009, 131, 455–458. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Etlinger, J.D.; Goldberg, A.L. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, T.; Kamada, Y.; Suzuki, K.; Kuboshima, N.; Akimatsu, H.; Ota, S.; Ohsumi, M.; Ohsumi, Y. Characterization of a novel autophagy-specific gene, ATG29. Biochem. Biophys. Res. Commun. 2005, 338, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Kawamata, T.; Suzuki, K.; Ohsumi, Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2007, 356, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Yorimitsu, T.; Reggiori, F.; Legakis, J.E.; Wang, C.-W.; Klionsky, D.J. Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell 2005, 16, 3438–3453. [Google Scholar] [CrossRef]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Chew, L.H.; Inoue-Aono, Y.; Cheong, H.; Nair, U.; Popelka, H.; Yip, C.K.; Klionsky, D.J. Atg29 phosphorylation regulates coordination of the Atg17-Atg31-Atg29 complex with the Atg11 scaffold during autophagy initiation. Proc. Natl. Acad. Sci. USA 2013, 110, E2875–E2884. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Tamura, N.; Kono, N.; Shimanaka, Y.; Arai, H.; Yamamoto, H.; Mizushima, N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017, 36, 1719–1735. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [Green Version]
- Schu, P.V.; Takegawa, K.; Fry, M.J.; Stack, J.H.; Waterfield, M.D.; Emr, S.D. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 1993, 260, 88–91. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Obara, K.; Sekito, T.; Niimi, K.; Ohsumi, Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 2008, 283, 23972–23980. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Mann, S.S.; Hammarback, J.A. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem. 1994, 269, 11492–11497. [Google Scholar]
- Sagiv, Y.; Legesse-Miller, A.; Porat, A.; Elazar, Z. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J. 2000, 19, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, H.; Vicini, S.; Olsen, R.W. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 11557–11562. [Google Scholar] [CrossRef] [Green Version]
- Legesse-Miller, A.; Sagiv, Y.; Glozman, R.; Elazar, Z. Aut7p, a soluble autophagic factor, participates in multiple membrane trafficking processes. J. Biol. Chem. 2000, 275, 32966–32973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Wu, Z.; Zhao, M.; Murtazina, R.; Cai, J.; Zhang, A.; Li, R.; Sun, D.; Li, W.; Zhao, L.; et al. Rab5-dependent autophagosome closure by ESCRT. J. Cell Biol. 2019, 218, 1908–1927. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Sánchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef]
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006, 281, 36303–36316. [Google Scholar] [CrossRef] [Green Version]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Nuttall, J.M.; Motley, A.M.; Hettema, E.H. Deficiency of the exportomer components Pex1, Pex6, and Pex15 causes enhanced pexophagy in Saccharomyces cerevisiae. Autophagy 2014, 10, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, M.; Francisco, T.; Lismont, C.; Hennebel, L.; Brees, C.; Wang, B.; Van Veldhoven, P.P.; Azevedo, J.E.; Fransen, M. Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy 2015, 11, 1326–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbé, S. Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018, 19, e45595. [Google Scholar] [CrossRef] [PubMed]
- Riccio, V.; Demers, N.; Hua, R.; Vissa, M.; Cheng, D.T.; Strilchuk, A.W.; Wang, Y.; McQuibban, G.A.; Kim, P.K. Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J. Cell Biol. 2019, 218, 798–807. [Google Scholar] [CrossRef] [Green Version]
- Bellu, A.R.; Salomons, F.A.; Kiel, J.A.K.W.; Veenhuis, M.; Van Der Klei, I.J. Removal of Pex3p is an important initial stage in selective peroxisome degradation in Hansenula polymorpha. J. Biol. Chem. 2002, 277, 42875–42880. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Abe, K.; Tatemichi, Y.; Fujiki, Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014, 10, 1549–1564. [Google Scholar] [CrossRef] [Green Version]
- Hara-Kuge, S.; Fujiki, Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp. Cell Res. 2008, 314, 3531–3541. [Google Scholar] [CrossRef]
- Farré, J.-C.; Manjithaya, R.; Mathewson, R.D.; Subramani, S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell 2008, 14, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Atg36: The Saccharomyces cerevisiae receptor for pexophagy. Autophagy 2012, 8, 1680–1681. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zientara-Rytter, K.; Ozeki, K.; Nazarko, T.Y.; Subramani, S. Pex3 and Atg37 compete to regulate the interaction between the pexophagy receptor, Atg30, and the Hrr25 kinase. Autophagy 2018, 14, 368–384. [Google Scholar] [CrossRef]
- Farré, J.-C.; Burkenroad, A.; Burnett, S.F.; Subramani, S. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 2013, 14, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, K.; Liu, X.; Feng, Y.; Klionsky, D.J. The progression of peroxisomal degradation through autophagy requires peroxisomal division. Autophagy 2014, 10, 652–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manivannan, S.; de Boer, R.; Veenhuis, M.; van der Klei, I.J. Lumenal peroxisomal protein aggregates are removed by concerted fission and autophagy events. Autophagy 2013, 9, 1044–1056. [Google Scholar] [CrossRef] [Green Version]
- Ano, Y.; Hattori, T.; Oku, M.; Mukaiyama, H.; Baba, M.; Ohsumi, Y.; Kato, N.; Sakai, Y. A sorting nexin PpAtg24 regulates vacuolar membrane dynamics during pexophagy via binding to phosphatidylinositol-3-phosphate. Mol. Biol. Cell 2005, 16, 446–457. [Google Scholar] [CrossRef]
- Wang, X.; Wang, P.; Zhang, Z.; Farré, J.-C.; Li, X.; Wang, R.; Xia, Z.; Subramani, S.; Ma, C. The autophagic degradation of cytosolic pools of peroxisomal proteins by a new selective pathway. Autophagy 2019, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, A.R.; Schandorff, S.; Høyer-Hansen, M.; Nielsen, M.O.; Jäättelä, M.; Dengjel, J.; Andersen, J.S. Ordered organelle degradation during starvation-induced autophagy. Mol. Cell. Proteom. 2008, 7, 2419–2428. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Hua, Z.; Mali, S.; McLoughlin, F.; Vierstra, R.D. ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell 2019, 177, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.-Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol. Cell 2019, 74, 330–346. [Google Scholar] [CrossRef] [Green Version]
- Turco, E.; Fracchiolla, D.; Martens, S. Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 2019, 19, 822–836. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kunau, W.H.; Hartig, A. Peroxisome biogenesis in Saccharomyces cerevisiae. Antonie Van Leeuwenhoek 1992, 62, 63–78. [Google Scholar] [CrossRef]
- Tuttle, D.L.; Lewin, A.S.; Dunn, W.A. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur. J. Cell Biol. 1993, 60, 283–290. [Google Scholar]
- Luiken, J.J.; van den Berg, M.; Heikoop, J.C.; Meijer, A.J. Autophagic degradation of peroxisomes in isolated rat hepatocytes. FEBS Lett. 1992, 304, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Iwata, J.I.; Ezaki, J.; Komatsu, M.; Yokota, S.; Ueno, T.; Tanida, I.; Chiba, T.; Tanaka, K.; Kominami, E. Excess Peroxisomes Are Degraded by Autophagic Machinery in Mammals. J. Biol. Chem. 2006, 281, 4035–4041. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Huybrechts, S.J.; Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome dynamics in cultured mammalian cells. Traffic 2009, 10, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.; Leighton, F.; De Duve, C. The synthesis and turnover of rat liver peroxisomes: II. Turnover of peroxisome proteins. J. Cell Biol. 1969, 41, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, V.E.; Sterling, W.R.; Tarantola, V.A.; Hartley, R.W.; Rechcigl, M. The kinetics of catalase synthesis and destruction in vivo. J. Biol. Chem. 1962, 237, 3468–3475. [Google Scholar] [PubMed]
- Burnett, S.F.; Farré, J.-C.; Nazarko, T.Y.; Subramani, S. Peroxisomal Pex3 activates selective autophagy of peroxisomes via interaction with the pexophagy receptor Atg30. J. Biol. Chem. 2015, 290, 8623–8631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarko, T.Y.; Ozeki, K.; Till, A.; Ramakrishnan, G.; Lotfi, P.; Yan, M.; Subramani, S. Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J. Cell Biol. 2014, 204, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Nazarko, T.Y. Atg37 regulates the assembly of the pexophagic receptor protein complex. Autophagy 2014, 10, 1348–1349. [Google Scholar] [CrossRef] [Green Version]
- Van Zutphen, T.; Veenhuis, M.; van der Klei, I.J. Damaged peroxisomes are subject to rapid autophagic degradation in the yeast Hansenula polymorpha. Autophagy 2011, 7, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1269. [Google Scholar] [CrossRef] [Green Version]
- Platta, H.W.; El Magraoui, F.; Bäumer, B.E.; Schlee, D.; Girzalsky, W.; Erdmann, R. Pex2 and pex12 function as protein-ubiquitin ligases in peroxisomal protein import. Mol. Cell. Biol. 2009, 29, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Van Zutphen, T.; Ciapaite, J.; Bloks, V.W.; Ackereley, C.; Gerding, A.; Jurdzinski, A.; de Moraes, R.A.; Zhang, L.; Wolters, J.C.; Bischoff, R.; et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J. Hepatol. 2016, 65, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; South, S.; Warren, D.; Jones, J.; Moser, A.B.; Moser, H.W.; Gould, S.J. Metabolic control of peroxisome abundance. J. Cell Sci. 1999, 112, 1579–1590. [Google Scholar] [PubMed]
- Nazarko, T.Y.; Farré, J.-C.; Subramani, S. Peroxisome size provides insights into the function of autophagy-related proteins. Mol. Biol. Cell 2009, 20, 3828–3839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibirny, A.A. Yeast peroxisomes: Structure, functions and biotechnological opportunities. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicot, A.-S.; Lo Verso, F.; Ratti, F.; Pilot-Storck, F.; Streichenberger, N.; Sandri, M.; Schaeffer, L.; Goillot, E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 2014, 10, 1036–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarko, V.Y.; Thevelein, J.M.; Sibirny, A.A. G-protein-coupled receptor Gpr1 and G-protein Gpa2 of cAMP-dependent signaling pathway are involved in glucose-induced pexophagy in the yeast Saccharomyces cerevisiae. Cell Biol. Int. 2008, 32, 502–504. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Futej, K.O.; Thevelein, J.M.; Sibirny, A.A. Differences in glucose sensing and signaling for pexophagy between the baker’s yeast Saccharomyces cerevisiae and the methylotrophic yeast Pichia pastoris. Autophagy 2008, 4, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Stasyk, O.V.; Stasyk, O.G.; Komduur, J.; Veenhuis, M.; Cregg, J.M.; Sibirny, A.A. A hexose transporter homologue controls glucose repression in the methylotrophic yeast Hansenula polymorpha. J. Biol. Chem. 2004, 279, 8116–8125. [Google Scholar] [CrossRef] [Green Version]
- Manjithaya, R.; Jain, S.; Farré, J.-C.; Subramani, S. A yeast MAPK cascade regulates pexophagy but not other autophagy pathways. J. Cell Biol. 2010, 189, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Wang, K.; Zhao, M.; Xu, T.; Klionsky, D.J. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J. Cell Biol. 2011, 193, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Lee, J.N.; Dutta, R.K.; Maharjan, Y.; Liu, Z.-Q.; Lim, J.-Y.; Kim, S.-J.; Cho, D.-H.; So, H.-S.; Choe, S.-K.; Park, R. Catalase inhibition induces pexophagy through ROS accumulation. Biochem. Biophys. Res. Commun. 2018, 501, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Eun, S.Y.; Lee, J.N.; Nam, I.-K.; Liu, Z.-Q.; So, H.-S.; Choe, S.-K.; Park, R. PEX5 regulates autophagy via the mTORC1-TFEB axis during starvation. Exp. Mol. Med. 2018, 50, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Han, H.; Zhou, M.-T.; Yang, B.; Ta, A.P.; Li, N.; Chen, J.; Wang, W. Proteomic Analysis of the Human Tankyrase Protein Interaction Network Reveals Its Role in Pexophagy. Cell Rep. 2017, 20, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Walter, C.; Gootjes, J.; Mooijer, P.A.; Portsteffen, H.; Klein, C.; Waterham, H.R.; Barth, P.G.; Epplen, J.T.; Kunau, W.H.; Wanders, R.J.; et al. Disorders of peroxisome biogenesis due to mutations in PEX1: Phenotypes and PEX1 protein levels. Am. J. Hum. Genet. 2001, 69, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defourny, J.; Aghaie, A.; Perfettini, I.; Avan, P.; Delmaghani, S.; Petit, C. Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl. Acad. Sci. USA 2019, 116, 8010–8017. [Google Scholar] [CrossRef] [Green Version]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Aksam, E.B.; Koek, A.; Kiel, J.A.K.W.; Jourdan, S.; Veenhuis, M.; van der Klei, I.J. A peroxisomal lon protease and peroxisome degradation by autophagy play key roles in vitality of Hansenula polymorpha cells. Autophagy 2007, 3, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabelo, N.; Martín, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Yakunin, E.; Moser, A.; Loeb, V.; Saada, A.; Faust, P.; Crane, D.I.; Baes, M.; Sharon, R. alpha-Synuclein abnormalities in mouse models of peroxisome biogenesis disorders. J. Neurosci. Res. 2010, 88, 866–876. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins Involved in Pexophagy | ||||
|---|---|---|---|---|
| Yeast | Mammalian | Function/Role | Reference | |
| Autophagosome Biogenesis | ||||
| Atg1p/ULK Complex | Atg1p | ULK1/2 | Ser/Thr kinase | [61] |
| Atg13p | ATG13 | Regulatory subunit of Atg1 (ULK1/2) complex | [63,65] | |
| Atg11p | Scaffold protein in pexophagic PAS | [68] | ||
| Atg17p | FIP200 | In complex with Atg29p-Atg31p (ATG13-ATG101) | [64,65,66] | |
| Atg29p | In complex with Atg17p and Atg31p | [62] | ||
| Atg31p | In complex with Atg17p and Atg29p | [68] | ||
| ATG101 | In complex with ATG13-FIP200 | [67] | ||
| Atg9p/ATG9 membrane cycling | Atg2p | ATG2 | Interacts with Atg18p (WIPI1/2) | [76] |
| Atg9p | ATG9A/B | Transmembrane protein; supplies membrane for autophagosome in vesicles | [87] | |
| Atg18p | WIPI1/2 | PtdIns3P-binding protein at autophagosome-ER contact sites | [76] | |
| PtdIns3K complex | Vps34p | VPS34 | PtdIns 3-kinase | [74,75] |
| Vps15p | VPS15 | Ser-Thr kinase | [74,75] | |
| Atg6p | BECN1 | Component of PtdIns3K complex I | [74,75] | |
| Atg14p | ATG14 | Component of PtdIns3K complex II | [74,75] | |
| Atg8p/LC3 Ubiquitin-like conjugation system | Atg8p | LC3A/B/C, GABARAP, GABARAPL1/2 | Ubiquitin-like protein conjugated to PE | [80,82,83,84,85] |
| Atg7p | ATG7 | E1-like enzyme | [80] | |
| Atg3p | ATG3 | E2-like enzyme | [80] | |
| Atg4p | ATG4A/B/C/D | Cysteine protease that cleaves Atg8 (LC3) | [81] | |
| Atg12p/ATG12 Ubiquitin-like conjugation system | Atg12p | ATG12 | Ubiquitin-like protein | [77] |
| Atg7p | ATG7 | E1-like enzyme | [77] | |
| Atg10p | ATG10 | E2-like enzyme | [78] | |
| Atg16p | ATG16L1 | Interacts with Atg5 and Atg12 (ATG5, ATG12) to aid ubiquitin-like conjugation | [79] | |
| Atg5p | ATG5 | Substrate of Atg12 (ATG12)-conjugation | [77] | |
| Peroxisome Designation | ||||
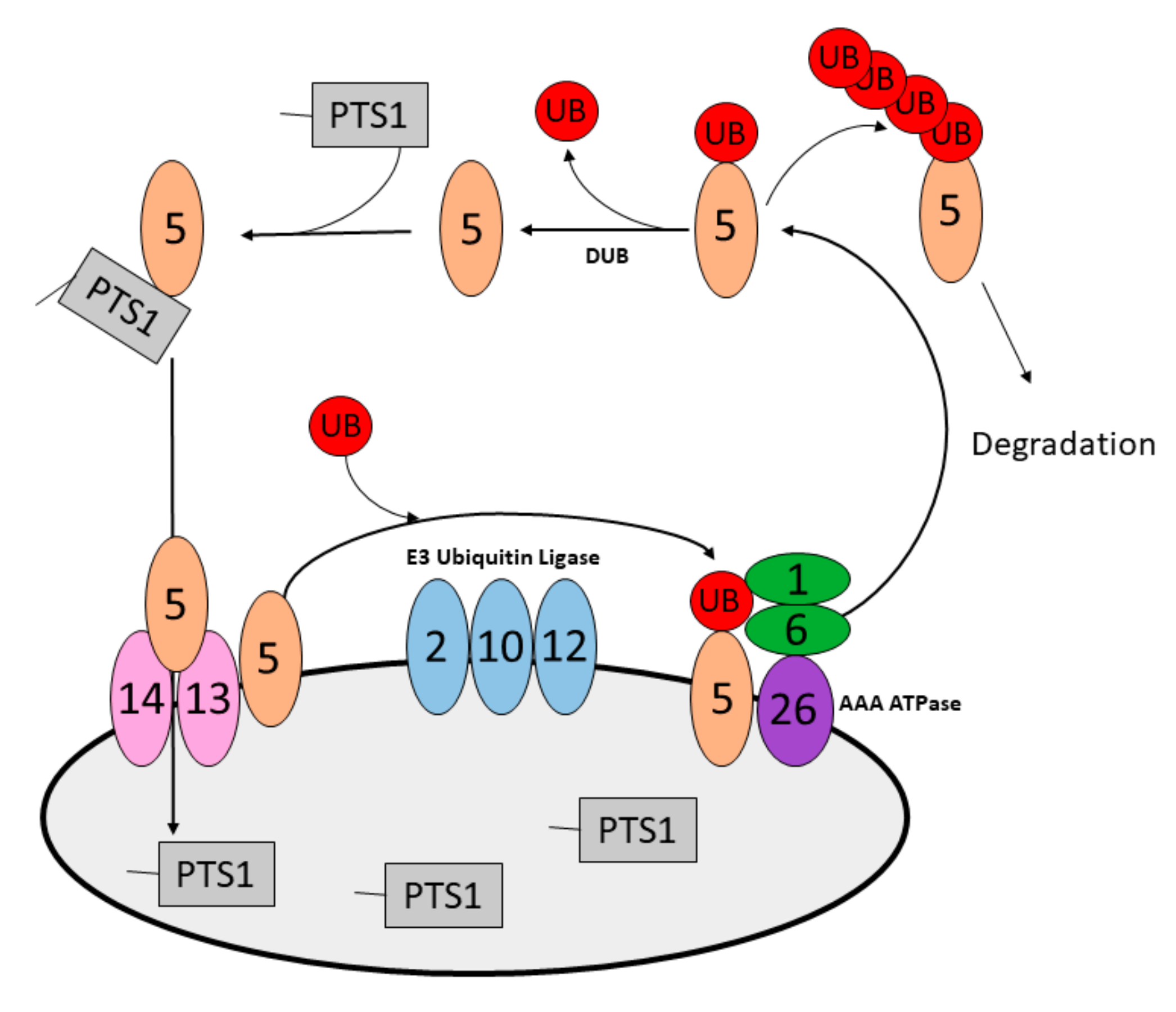
| AAA-type ATPase Complex | Pex1p | PEX1 | In complex with Pex6-Pex15 (PEX6-PEX26); Pex5 (PEX5) receptor recycling; defects signal pexophagy | [99,100] |
| Pex6p | PEX6 | In complex with Pex1-Pex15 (PEX1-PEX26) | [99,100] | |
| Pex15p | PEX26 | In complex with Pex1-Pex6 (PEX1-PEX6) | [99,100] | |
| E3 Ubiquitin Ligase | PEX2 | In complex with PEX10-PEX12; ubiquitinates PEX5 and PMP70 to signal mammalian pexophagy | [101] | |
| Ubiquitin Targets | PEX5 | Matrix protein import receptor; accumulated PEX5-UBB signals pexophagy | [99,100,101,102] | |
| PMP70 | PMP; accumulated PMP70-UBB signals pexophagy | [101] | ||
| Deubiquitinase | USP30 | DUB; removes ubiquitin from PEX5 and PMP70 to oppose pexophagy | [103,104] | |
| Unknown | Pex3p | PEX3 | Biogenesis factor; loss signals yeast pexophagy, over-expression signals mammalian pexophagy | [105,106] |
| Pex14p | PEX14 | Defects signal yeast pexophagy; potential signal for mammalian pexophagy | [99,107] | |
| Peroxisome Targeting and Sequestration | ||||
| Pexophagy Receptors | Atg30p | Links peroxisomes to autophagy machinery; P. pastoris | [108] | |
| Atg36p | Links peroxisomes to autophagy machinery; S. cerevisiae | [109,110] | ||
| NBR1 | Links peroxisomes to autophagy machinery; primary receptor | [111,112] | ||
| p62 | Links peroxisomes to autophagy machinery; enhances NBR1-mediated pexophagy | [111,112] | ||
| Receptor Ligands | Atg37p | ACBD5 | Tethers Atg30p to peroxisomes in P. pastoris; regulates formation of receptor protein complex | [113] |
| Pex3p | Binds and tethers Atg30p to peroxisomes in P. pastoris; and Atg36p to peroxisomes in S. cerevisiae | [114] | ||
| UBB | NBR1 and p62 bind ubiquitin via UBA | [111] | ||
| Kinases | Hrr25p | Phosphorylates Atg30p in P. pastoris and Atg36p in S. cerevisiae; promotes pexophagy | [113,114] | |
| Fission Machinery | Dnm1p | Fission machinery; pinches off peroxisomes for yeast pexophagy | [115,116] | |
| Vps1p | ||||
| Fis1p | ||||
| Mffp | ||||
| Pex11p | ||||
| Peroxisome Degradation | ||||
| Fusion Machinery | Atg24p | PtdIns3P-binding protein; required for vacuolar fusion in pexophagy | [117] | |
| Cytosolic Peroxin Degradation | Atg11p | Facilitates degradation of Pex5p and Pex7p during pexophagy | [118] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. https://doi.org/10.3390/ijms21020578
Germain K, Kim PK. Pexophagy: A Model for Selective Autophagy. International Journal of Molecular Sciences. 2020; 21(2):578. https://doi.org/10.3390/ijms21020578
Chicago/Turabian StyleGermain, Kyla, and Peter K. Kim. 2020. "Pexophagy: A Model for Selective Autophagy" International Journal of Molecular Sciences 21, no. 2: 578. https://doi.org/10.3390/ijms21020578
APA StyleGermain, K., & Kim, P. K. (2020). Pexophagy: A Model for Selective Autophagy. International Journal of Molecular Sciences, 21(2), 578. https://doi.org/10.3390/ijms21020578