Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges


Abstract
:1. Introduction
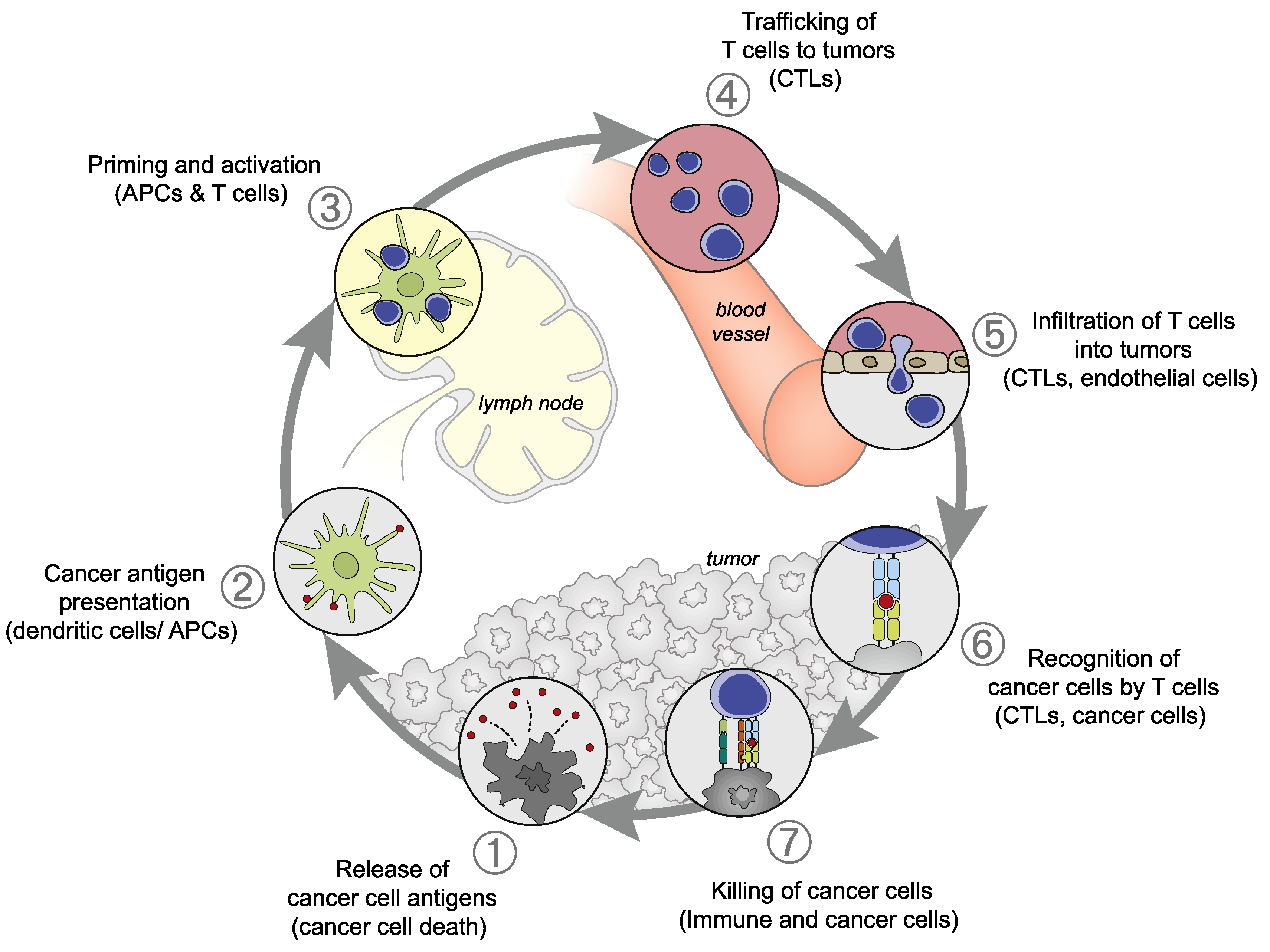
2. Cancer-Immunity Cycle
2.1. Cancer-Immunity Cycle Outlined
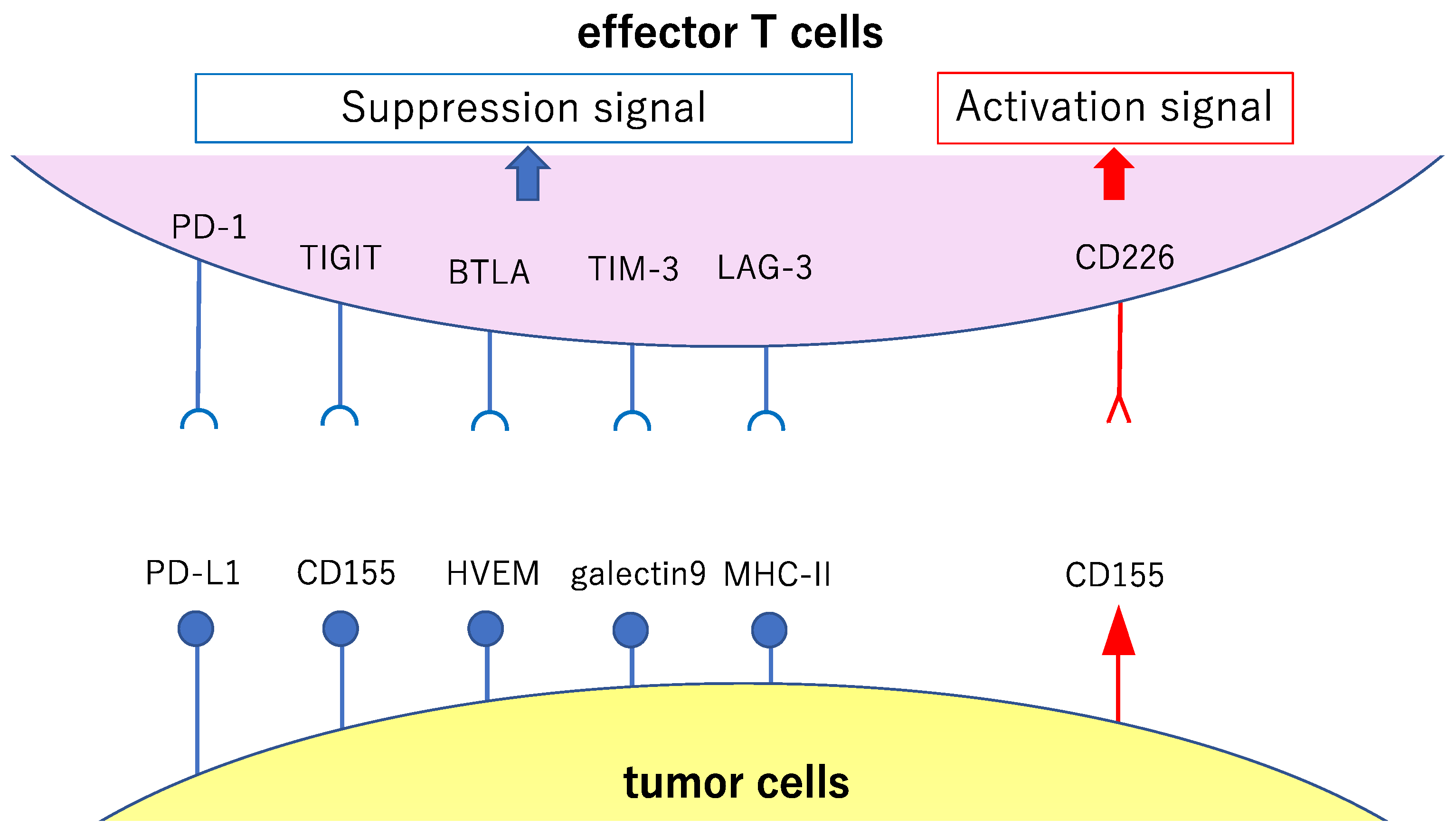
2.2. CD8-Positive Cytotoxic T Cells
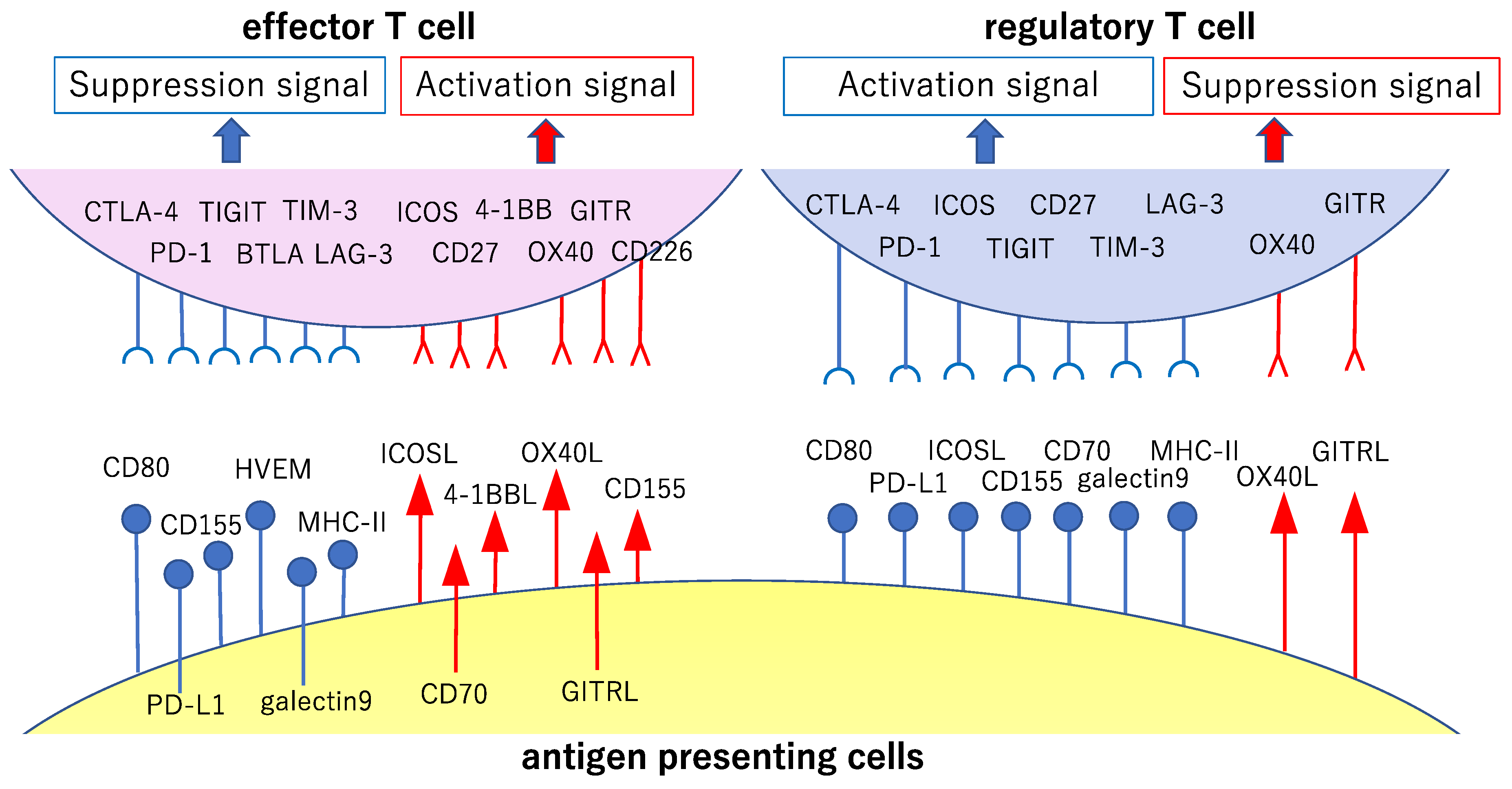
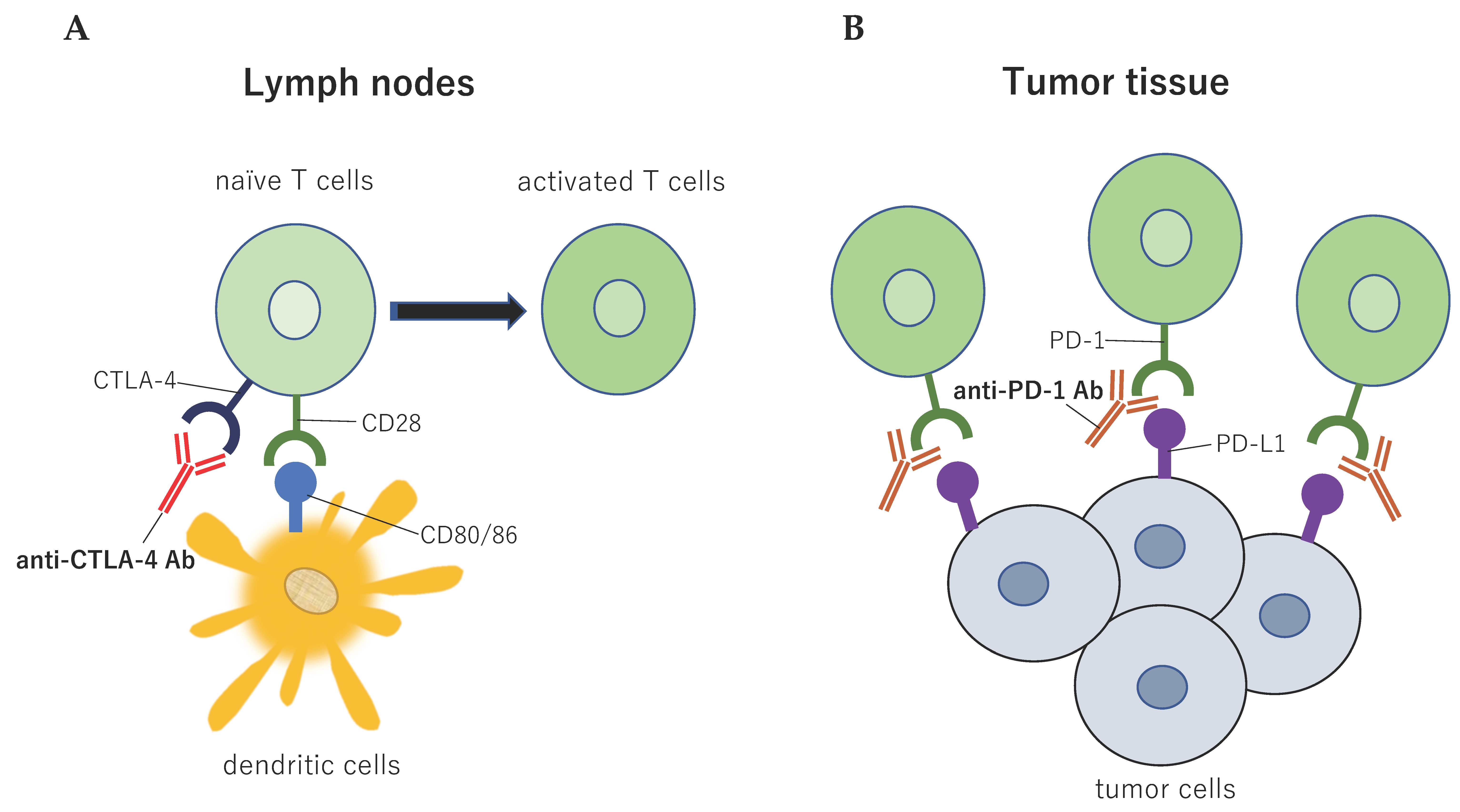
2.3. CD28 Family: Cytotoxic T-lymphocyte Associated Protein 4 and Programmed Cell Death 1
2.4. TNF Receptor Superfamily: OX-40 and Glucocorticoid-Induced TNFR-Related (GITR) Gene
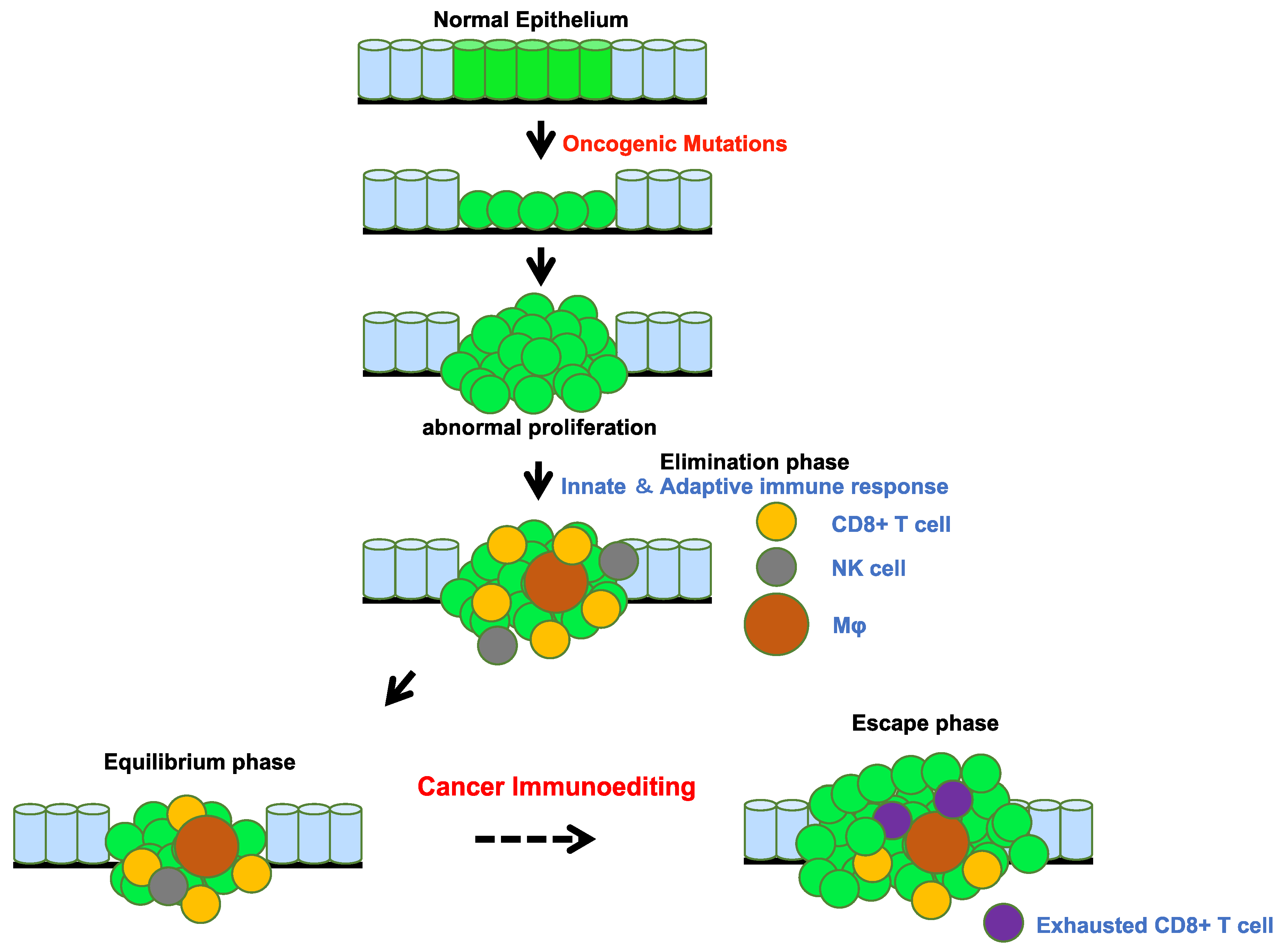
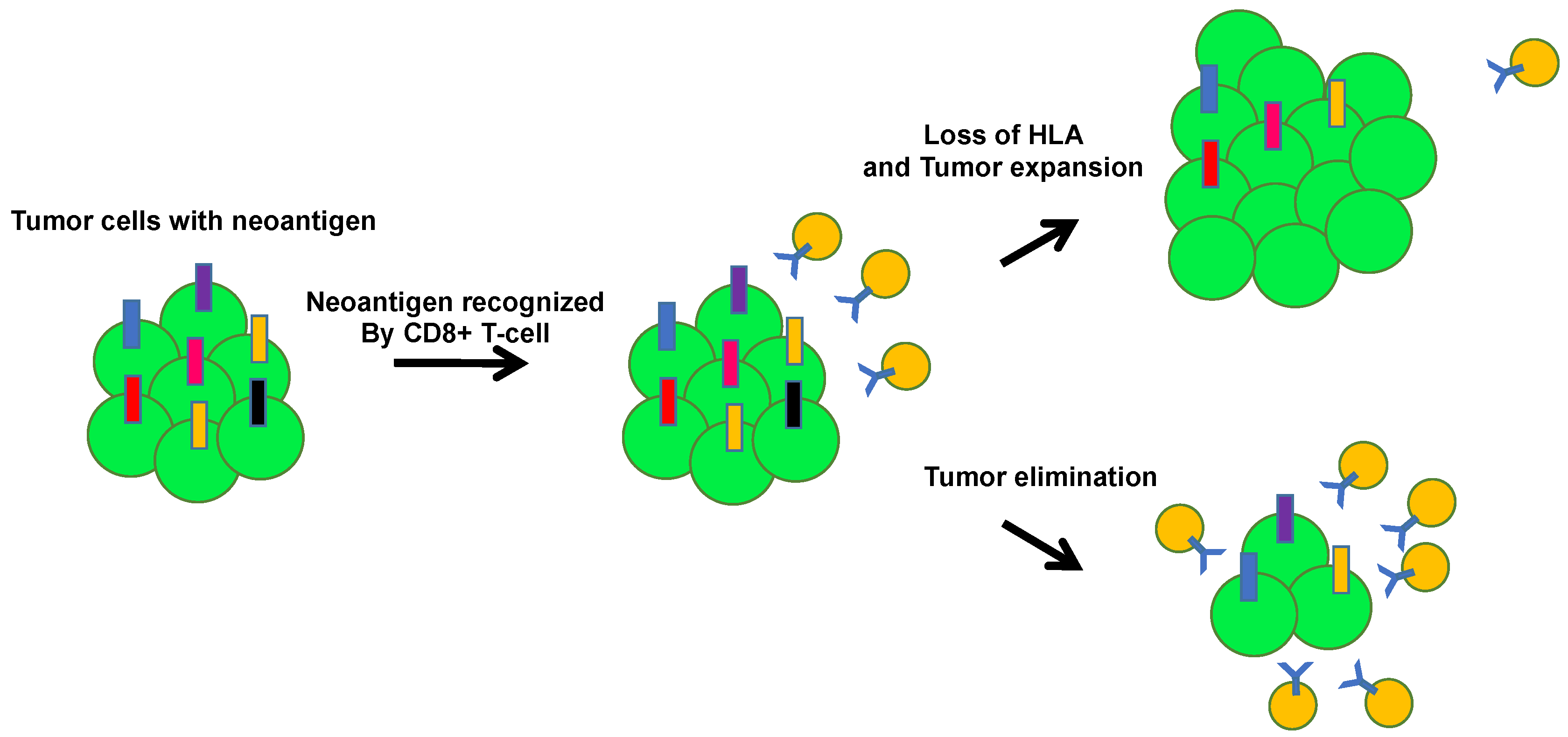
3. Tumors Escape Immunosurveillance through Immunoediting
4. Human Leukocyte Antigen (HLA) Loss and Immune Escape in Lung Cancer Evolution
5. Heterogenous Immunoediting in Lung Cancer
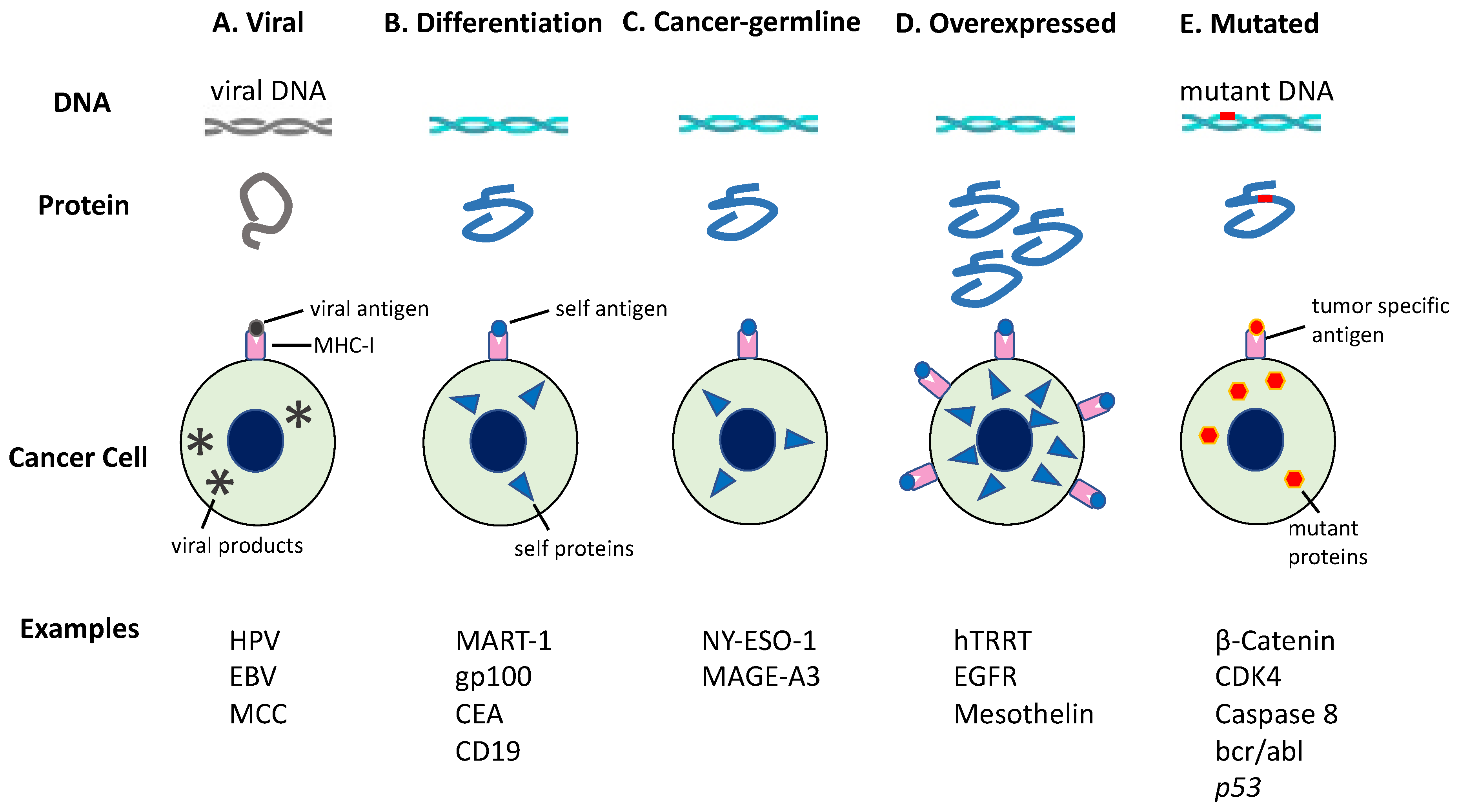
6. Neoantigen Derived from Mutation
6.1. Tumor Antigens
6.2. Anti-Tumor Immune Responses by Neoantigen-Specific T Cells
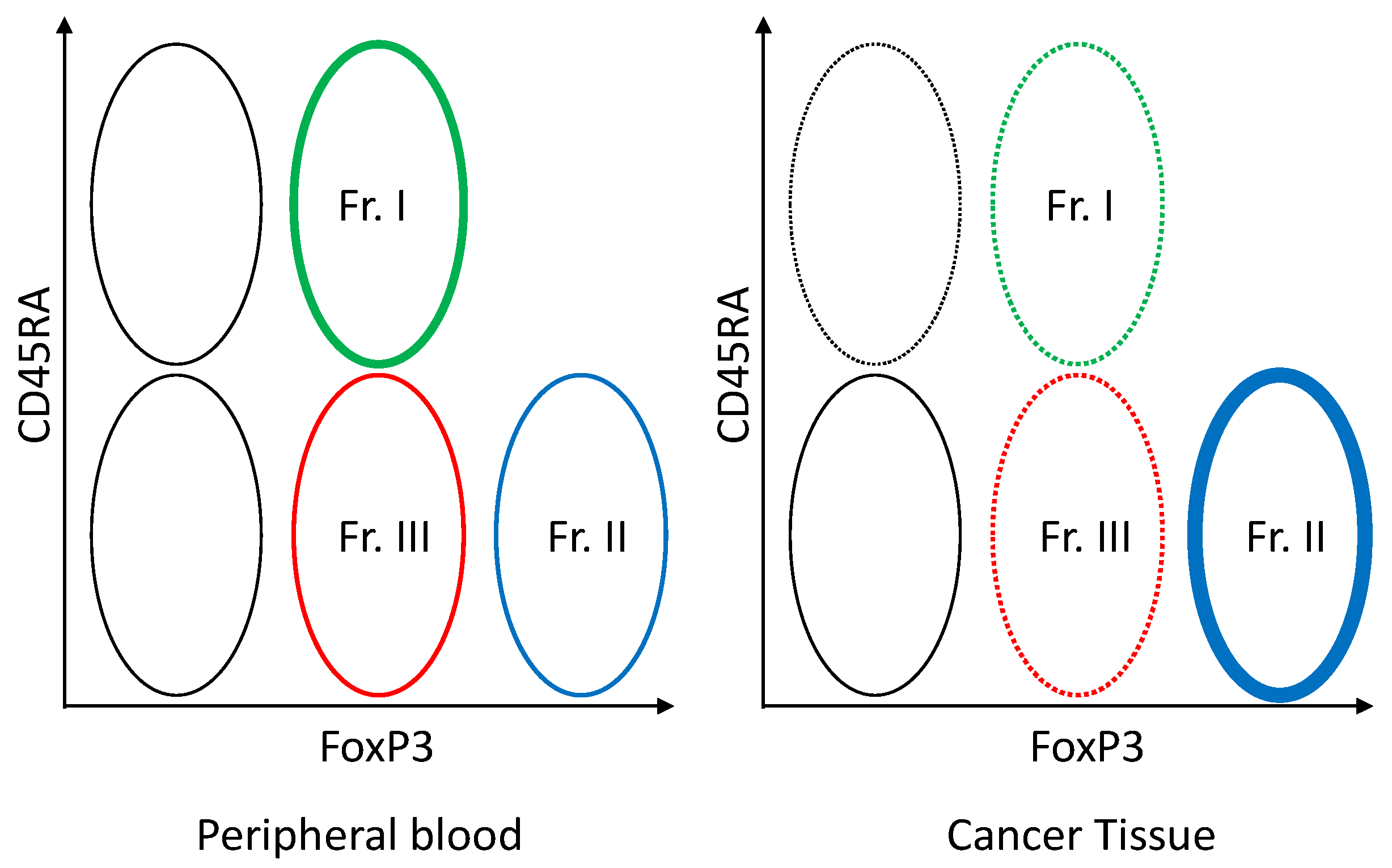
7. Treg and Tumor Immunity
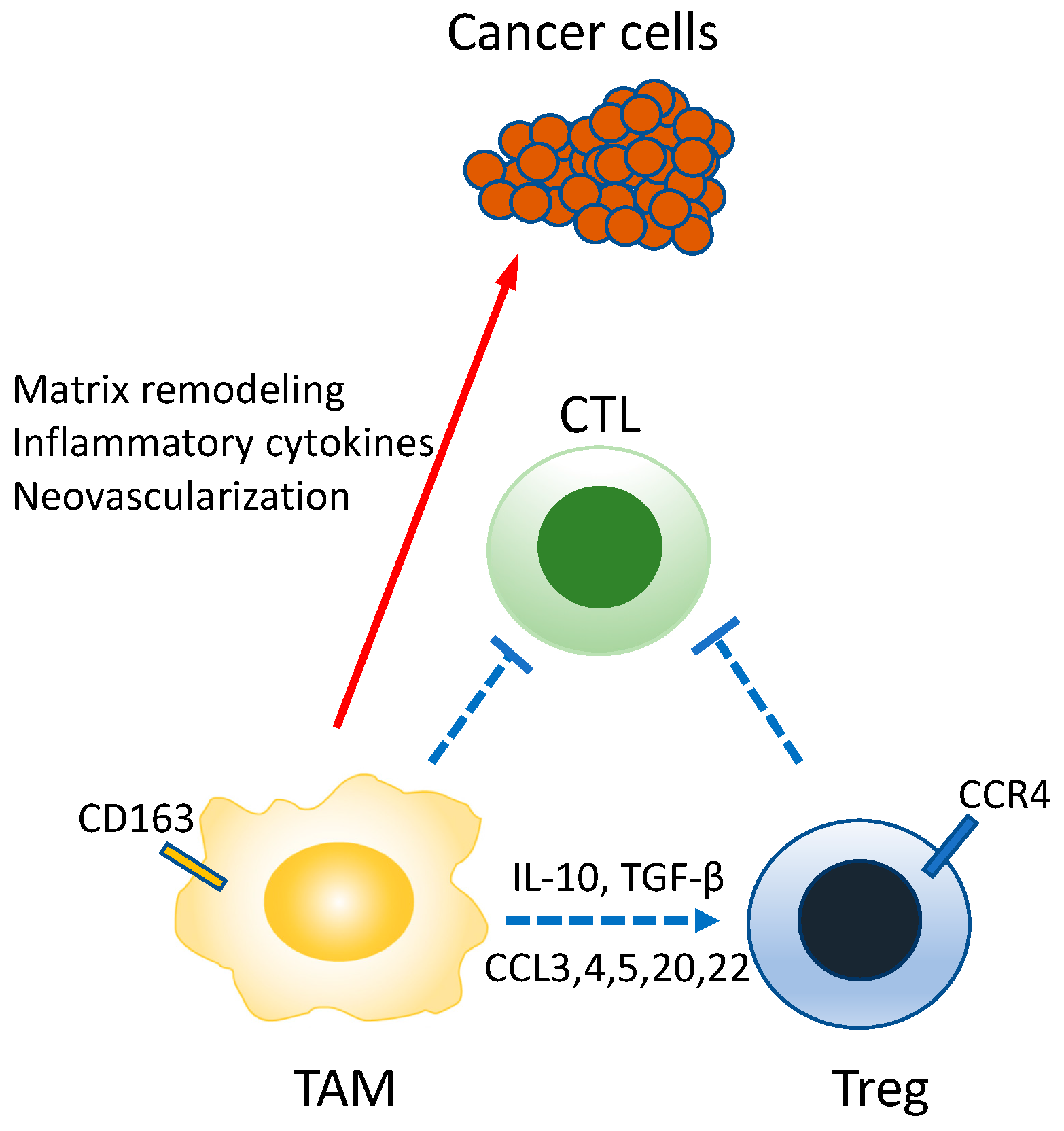
8. Tumor-Associated Macrophages (TAMs) and Tumor Immunity
9. Methods of Overcoming Tumor Immunoediting
9.1. Success of Immune Checkpoint Inhibitors in NSCLC Patients
9.2. Treg-Targeting Treatment
9.3. TAM-Targeting Cancer Treatment
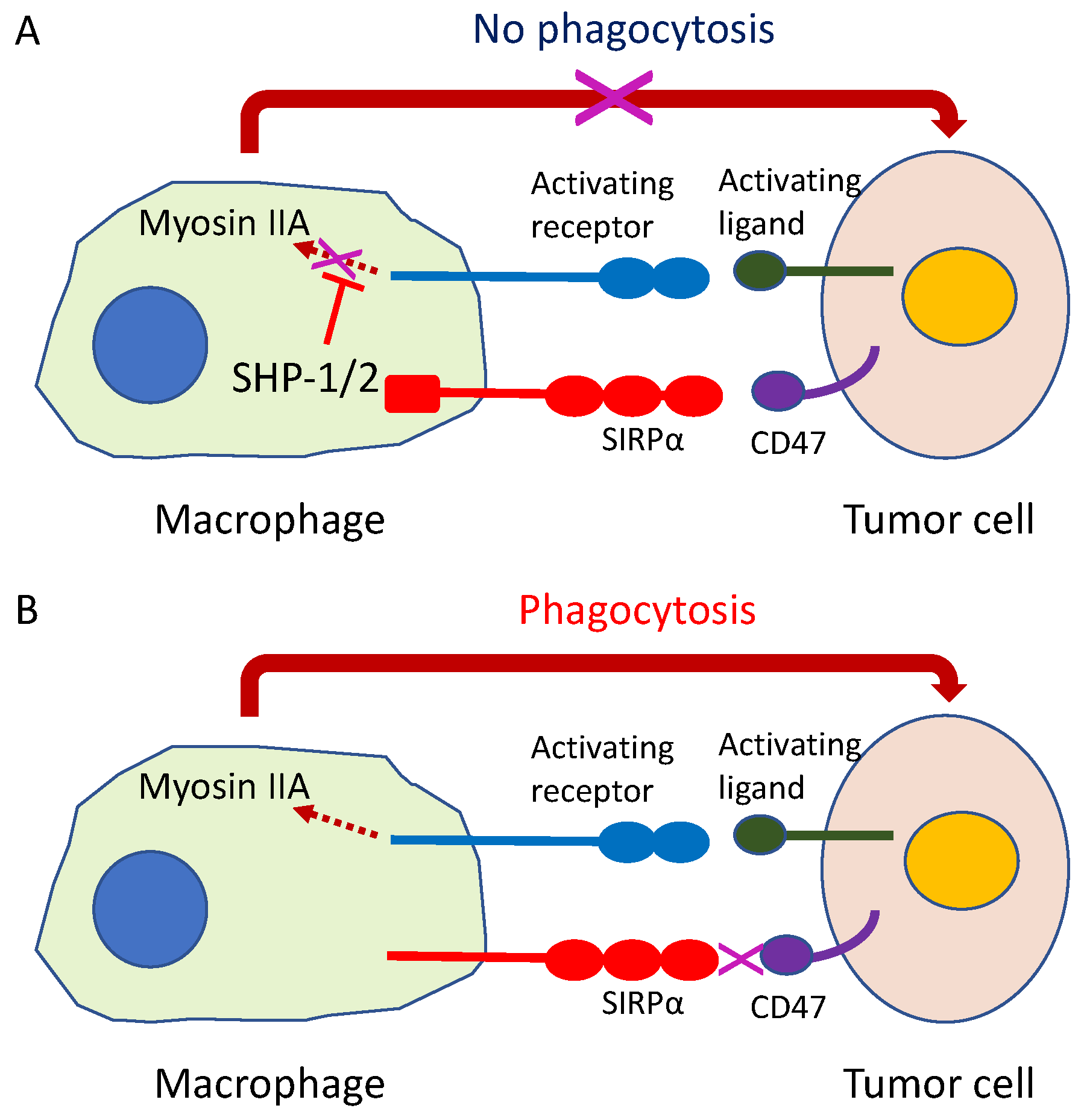
9.4. Phagocytosis Checkpoint Inhibitors
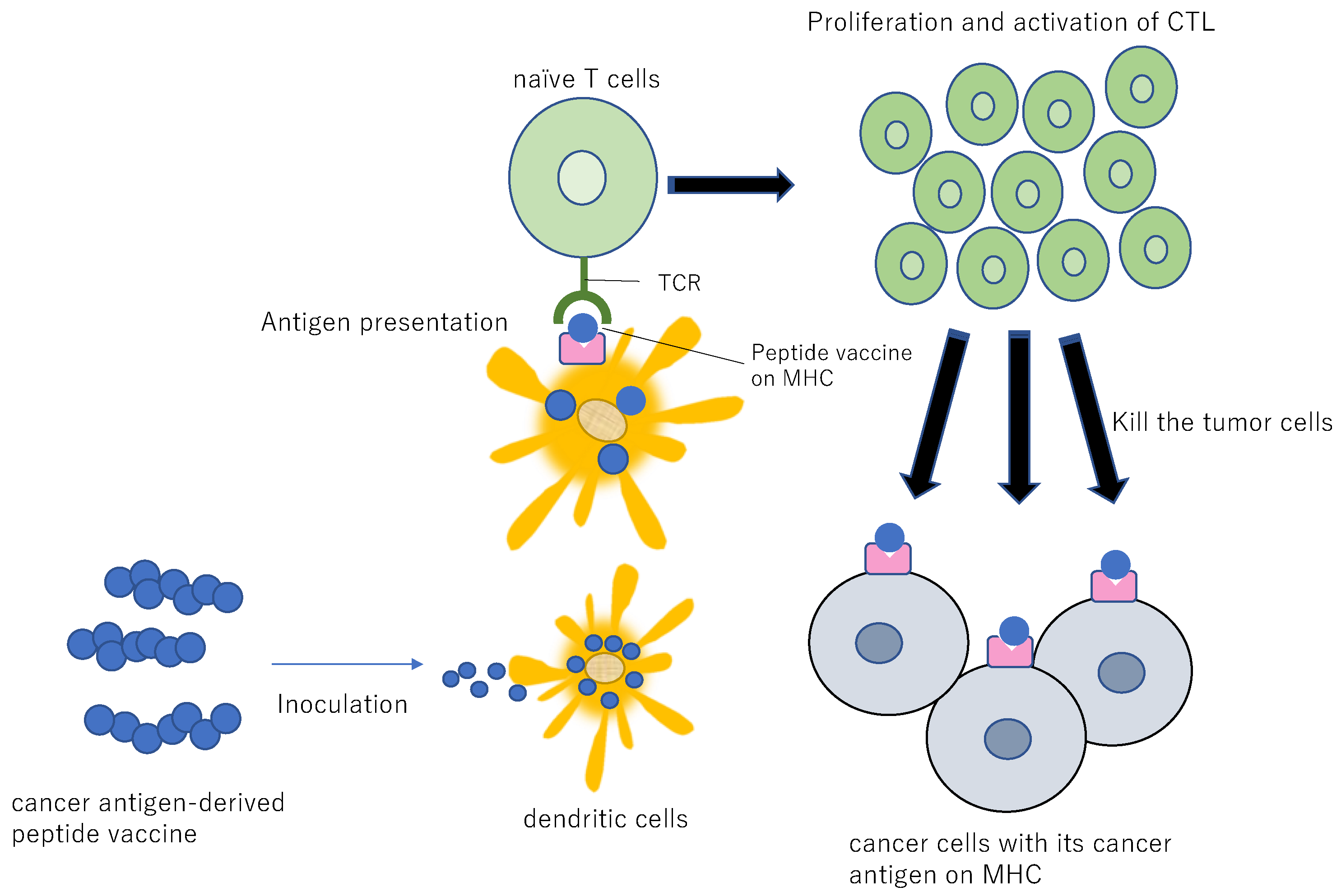
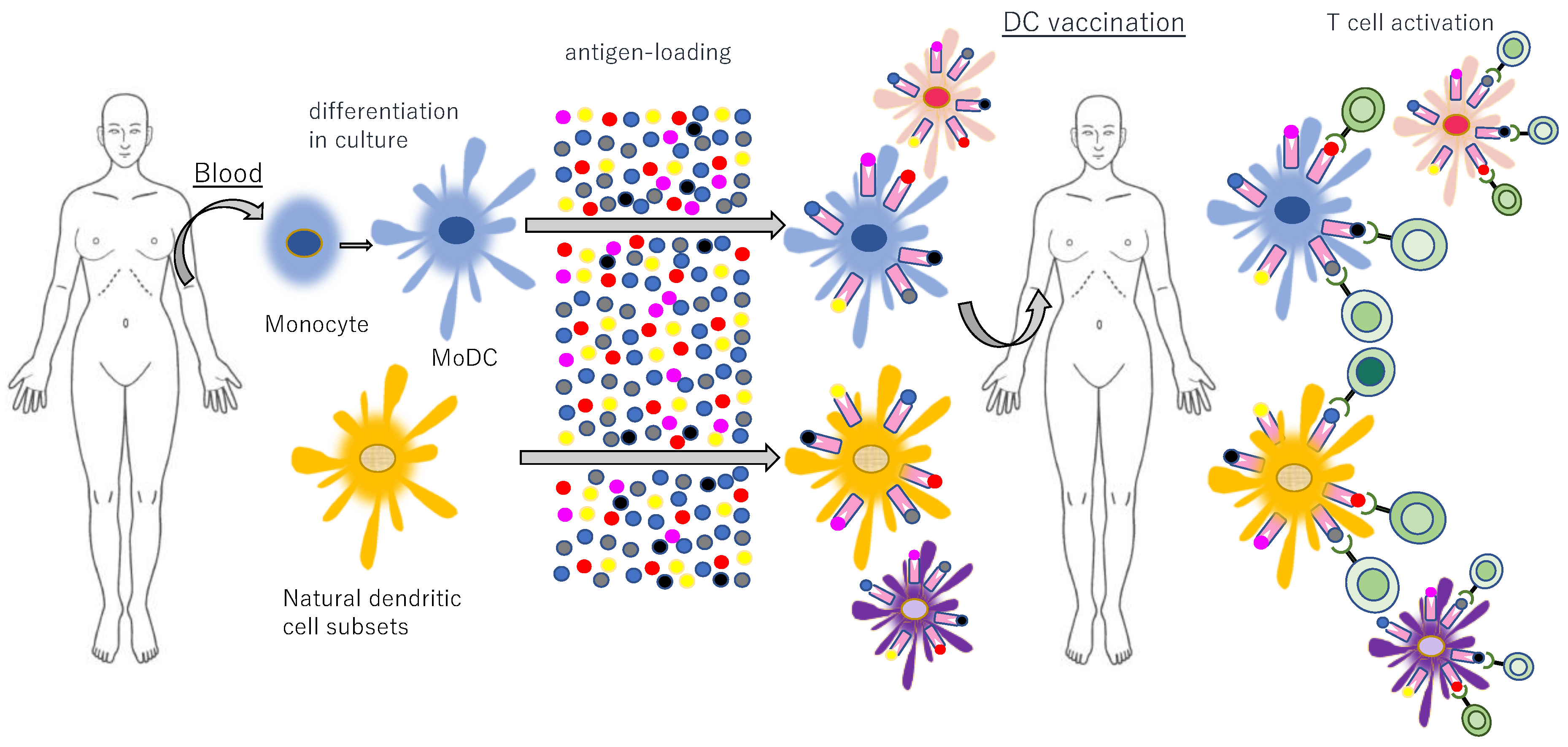
9.5. Cancer Vaccines
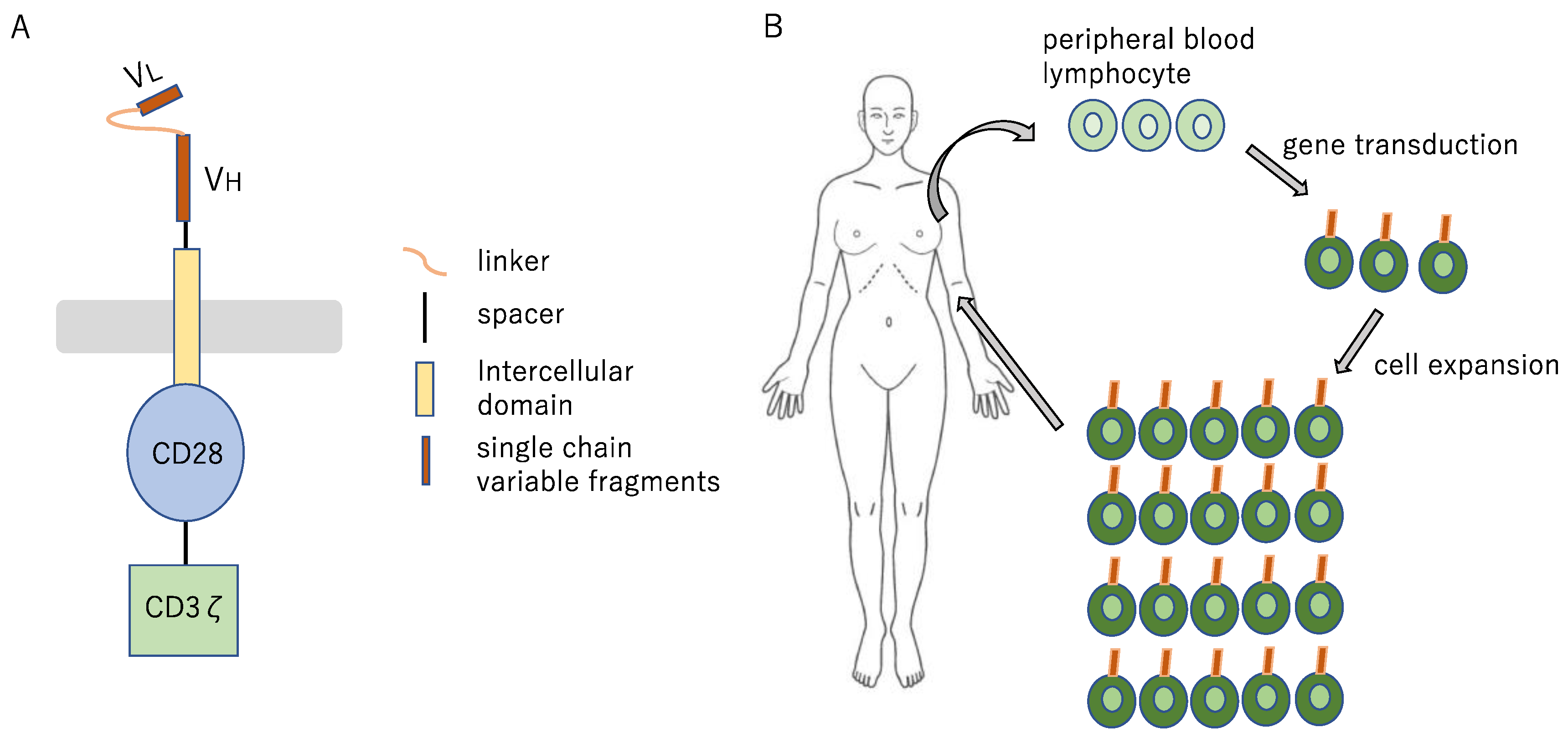
9.6. Chimeric Antigen Receptor T Cell and Bispecific Antibodies for Lung Cancer
10. Future Perspective
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MHC I | Major histocompatibility complex class I |
| PD-L1 | Programmed cell death ligand 1 |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| NK | Natural killer |
| HLA | Human leukocyte antigen |
| LOH | Loss of heterozygosity |
| NSCLCs | Non-small-cell lung cancers |
| TCGA | The Cancer Genome Atlas |
| TRACERx | TRAcking Cancer Evolution through Therapy (Rx) |
| TCR | T cell antigen receptor |
| ICIs | Immune checkpoint inhibitors |
| Ig | Immunoglobulin |
| OS | Overall survival |
| PFS | Progression-free survival |
| TMB | Tumor mutation burden |
| APCs | Antigen-presenting cells |
| CD47-SIRPα | CD47-signal-regulatory protein α |
| DCs | Dendritic cells |
| FLT3L | FMS-like tyrosine kinase 3 ligand |
| TGFβ | Transforming growth factor- β |
| CCL | CC-chemokine ligand |
| VEGF | Vascular endothelial growth factor |
| NY-ESO-1 | New York esophageal squamous cell carcinoma 1 |
| TILs | Tumor-infiltrating lymphocytes |
| CAR | Chimeric antigen receptor |
References
- Nauts, H.C.; Swift, W.E.; Coley, B.L. The treatment of malignant tumors by bacterial toxins as developed by the late William B. Coley, M.D., reviewed in the light of modern research. Cancer Res. 1946, 6, 205–216. [Google Scholar]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Krause, R.M. Paul Ehrlich and O.T. Avery: Pathfinders in the search for immunity. Vaccine 1999, 17 (Suppl. 3), S64–S67. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Engel, A.M.; Thomsen, A.R.; Werdelin, O. The susceptibility to cytotoxic T lymphocyte mediated lysis of chemically induced sarcomas from immunodeficient and normal mice. Scand. J. Immunol. 1997, 45, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, R.; Goto, T.; Hirotsu, Y.; Nakagomi, T.; Yokoyama, Y.; Otake, S.; Amemiya, K.; Oyama, T.; Omata, M. PD-L1 Expression and Tumor-Infiltrating Lymphocytes in Thymic Epithelial Neoplasms. J. Clin. Med. 2019, 8, 1833. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Callahan, M.K.; Postow, M.A.; Wolchok, J.D. On being less tolerant: Enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med. 2015, 7, 280–281. [Google Scholar] [CrossRef]
- Pentcheva-Hoang, T.; Corse, E.; Allison, J.P. Negative regulators of T-cell activation: Potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol. Rev. 2009, 229, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Wolchok, J.D.; Allison, J.P. Anti-CTLA-4 antibody therapy: Immune monitoring during clinical development of a novel immunotherapy. Semin. Oncol. 2010, 37, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Blank, C.; Kuball, J.; Voelkl, S.; Wiendl, H.; Becker, B.; Walter, B.; Majdic, O.; Gajewski, T.F.; Theobald, M.; Andreesen, R.; et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses In Vitro. Int. J. Cancer 2006, 119, 317–327. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Duan, W.; Choi, H.; Eun, S.Y.; Madireddi, S.; Mehta, A. TNF superfamily in inflammatory disease: Translating basic insights. Trends Immunol. 2012, 33, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Hirschhorn-Cymerman, D.; Morales-Kastresana, A.; Sanmamed, M.F.; Wolchok, J.D. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin. Cancer Res. 2013, 19, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, A.; Fujii, N.; Murakami, T.; Kiura, K.; Aoe, K.; Yamane, H.; Masuda, K.; Maeda, T.; Sugi, K.; Darzynkiewicz, Z.; et al. High frequency of allele-specific down-regulation of HLA class I expression in lung cancer cell lines. Anticancer Res. 2004, 24, 1525–1528. [Google Scholar]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, A.A.; Yu, T.F.; Leach, D.R.; Allison, J.P. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 10067–10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Kenter, G.G.; Ferrone, S.; Fleuren, G.J. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol. Immunother. 2008, 57, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Klippel, Z.K.; Chou, J.; Towlerton, A.M.; Voong, L.N.; Robbins, P.; Bensinger, W.I.; Warren, E.H. Immune escape from NY-ESO-1-specific T-cell therapy via loss of heterozygosity in the MHC. Gene Ther. 2014, 21, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Esch, E.M.; Tummers, B.; Baartmans, V.; Osse, E.M.; Ter Haar, N.; Trietsch, M.D.; Hellebrekers, B.W.; Holleboom, C.A.; Nagel, H.T.; Tan, L.T.; et al. Alterations in classical and nonclassical HLA expression in recurrent and progressive HPV-induced usual vulvar intraepithelial neoplasia and implications for immunotherapy. Int. J. Cancer 2014, 135, 830–842. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Cancer Evolution Constrained by the Immune Microenvironment. Cell 2017, 170, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Joshi, K.; Robert de Massy, M.; Ismail, M.; Reading, J.L.; Uddin, I.; Woolston, A.; Hatipoglu, E.; Oakes, T.; Rosenthal, R.; Peacock, T.; et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019, 25, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Otake, S.; Goto, T. Stereotactic Radiotherapy for Oligometastasis. Cancers 2019, 11, 133. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Labiano, S.; Palazon, A.; Melero, I. Immune response regulation in the tumor microenvironment by hypoxia. Semin. Oncol. 2015, 42, 378–386. [Google Scholar] [CrossRef]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, M.; Miedema, D.M.; Waclaw, B.; Veenstra, V.L.; Lecca, M.C.; Nijman, L.E.; van Dijk, E.; van Neerven, S.M.; Lodestijn, S.C.; Lenos, K.J.; et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. USA 2019, 116, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, S.; Pritchard, A.L. Identifying neoantigens for use in immunotherapy. Mamm. Genome 2018, 29, 714–730. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. BioMed Res. Int. 2015, 2015, 948501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T. Radiation as an In Situ Auto-Vaccination: Current Perspectives and Challenges. Vaccines 2019, 7, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloosen, S.; Arnold, J.; Thio, M.; Bos, G.M.; Kyewski, B.; Germeraad, W.T. Expression of tumor-associated differentiation antigens, MUC1 glycoforms and CEA, in human thymic epithelial cells: Implications for self-tolerance and tumor therapy. Cancer Res. 2007, 67, 3919–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksic, M.; Liddy, N.; Molloy, P.E.; Pumphrey, N.; Vuidepot, A.; Chang, K.M.; Jakobsen, B.K. Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol. 2012, 42, 3174–3179. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.P.; Gerry, A.B.; Brewer, J.E.; Melchiori, L.; Bridgeman, J.S.; Bennett, A.D.; Pumphrey, N.J.; Jakobsen, B.K.; Price, D.A.; Ladell, K.; et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin. Exp. Immunol. 2015, 180, 255–270. [Google Scholar] [CrossRef] [Green Version]
- Nakagomi, T.; Goto, T.; Hirotsu, Y.; Shikata, D.; Yokoyama, Y.; Higuchi, R.; Otake, S.; Amemiya, K.; Oyama, T.; Mochizuki, H.; et al. Genomic Characteristics of Invasive Mucinous Adenocarcinomas of the Lung and Potential Therapeutic Targets of B7-H3. Cancers 2018, 10, 478. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Robbins, P.F. Targeting neoantigens for cancer immunotherapy. Int. Immunol. 2016, 28, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Hirotsu, Y.; Mochizuki, H.; Nakagomi, T.; Shikata, D.; Yokoyama, Y.; Oyama, T.; Amemiya, K.; Okimoto, K.; Omata, M. Mutational analysis of multiple lung cancers: Discrimination between primary and metastatic lung cancers by genomic profile. Oncotarget 2017, 8, 31133–31143. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Fotakis, G.; Rieder, D.; Haider, M.; Trajanoski, Z.; Finotello, F. NeoFuse: Predicting fusion neoantigens from RNA sequencing data. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Galuppini, F.; Dal Pozzo, C.A.; Deckert, J.; Loupakis, F.; Fassan, M.; Baffa, R. Tumor mutation burden: From comprehensive mutational screening to the clinic. Cancer Cell Int. 2019, 19, 209. [Google Scholar] [CrossRef]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2019. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Nejo, T.; Matsushita, H.; Karasaki, T.; Nomura, M.; Saito, K.; Tanaka, S.; Takayanagi, S.; Hana, T.; Takahashi, S.; Kitagawa, Y.; et al. Reduced Neoantigen Expression Revealed by Longitudinal Multiomics as a Possible Immune Evasion Mechanism in Glioma. Cancer Immunol. Res. 2019, 7, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Aragon-Sanabria, V.; Kim, G.B.; Dong, C. From Cancer Immunoediting to New Strategies in Cancer Immunotherapy: The Roles of Immune Cells and Mechanics in Oncology. Adv. Exp. Med. Biol. 2018, 1092, 113–138. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Kisielow, P. How does the immune system learn to distinguish between good and evil? The first definitive studies of T cell central tolerance and positive selection. Immunogenetics 2019, 71, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Kim, J.H.; Falo, L.D., Jr.; You, Z. Tumor regulatory T cells potently abrogate antitumor immunity. J. Immunol. 2009, 182, 6160–6167. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, D.A.; Fahmy, T.M.; Piccirillo, C.A.; La Cava, A. Rebalancing Immune Homeostasis to Treat Autoimmune Diseases. Trends Immunol. 2019, 40, 888–908. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Verma, N.D.; Robinson, C.M.; Carter, N.; Wilcox, P.; Tran, G.T.; Wang, C.; Sharland, A.; Nomura, M.; Plain, K.M.; Bishop, G.A.; et al. Alloactivation of Naive CD4(+)CD8(-)CD25(+)T Regulatory Cells: Expression of CD8alpha Identifies Potent Suppressor Cells That Can Promote Transplant Tolerance Induction. Front. Immunol. 2019, 10, 2397. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T. Regulatory T cells: How do they suppress immune responses? Int. Immunol. 2009, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Kishi, A.; Osaki, M.; Morikawa, H.; Prieto-Martin, P.; Wing, K.; Saito, T.; Sakaguchi, S. Construction of self-recognizing regulatory T cells from conventional T cells by controlling CTLA-4 and IL-2 expression. Proc. Natl. Acad. Sci. USA 2013, 110, E2116–E2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosse, M.E.; Nederlof, I.; Walker, L.S.K.; Samsom, J.N. Tipping the balance: Inhibitory checkpoints in intestinal homeostasis. Mucosal Immunol. 2019, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.F.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Regulatory T cells in melanoma: The final hurdle towards effective immunotherapy? Lancet Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [Green Version]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 2008, 105, 10113–10118. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Hawley, C.A.; Garner, H.; Scott, C.L.; Schridde, A.; Steers, N.J.; Mack, M.; Joshi, A.; Guilliams, M.; Mowat, A.M.; et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat. Commun. 2016, 7, ncomms11852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, A.; Szenajch, J.; Ostrowska, G.; Wojtowicz, A.; Wojtowicz, K.; Kruszewski, A.A.; Maruszynski, M.; Aukerman, S.L.; Wiktor-Jedrzejczak, W. Impaired tumor growth in colony-stimulating factor 1 (CSF-1)-deficient, macrophage-deficient op/op mouse: Evidence for a role of CSF-1-dependent macrophages in formation of tumor stroma. Int. J. Cancer 1996, 65, 112–119. [Google Scholar] [CrossRef]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yang, L.; Yu, L.; Wang, Y.Y.; Chen, R.; Qian, J.; Hong, Z.P.; Su, X.S. Surgery-induced monocytic myeloid-derived suppressor cells expand regulatory T cells in lung cancer. Oncotarget 2017, 8, 17050–17058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. BioMed Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Invest. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; Macrae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Bashir, B.; Wilson, M.A. Novel Immunotherapy Combinations. Curr. Oncol. Rep. 2019, 21, 96. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Pardoll, D.M. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 379, e14. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Borghaei, H.; Ramalingam, S.S.; Horn, L.; De Castro Carpeno, J.; Pluzanski, A.; Burgio, M.A.; Garassino, M.; Chow, L.Q.M.; Gettinger, S.; et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: A pooled analysis. Lancet Oncol. 2019, 20, 1395–1408. [Google Scholar] [CrossRef]
- Tseng, C.W.; Hung, C.F.; Alvarez, R.D.; Trimble, C.; Huh, W.K.; Kim, D.; Chuang, C.M.; Lin, C.T.; Tsai, Y.C.; He, L.; et al. Pretreatment with cisplatin enhances E7-specific CD8+ T-Cell-mediated antitumor immunity induced by DNA vaccination. Clin. Cancer Res. 2008, 14, 3185–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Gandhi, L.; Garassino, M.C. Pembrolizumab plus Chemotherapy in Lung Cancer. N. Engl. J. Med. 2018, 379, e18. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Walunas, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindan, R.; Szczesna, A.; Ahn, M.J.; Schneider, C.P.; Gonzalez Mella, P.F.; Barlesi, F.; Han, B.; Ganea, D.E.; Von Pawel, J.; Vladimirov, V.; et al. Phase III Trial of Ipilimumab Combined With Paclitaxel and Carboplatin in Advanced Squamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2017, 35, 3449–3457. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.; Goldberg, S.B.; Balmanoukian, A.; Chaft, J.E.; Sanborn, R.E.; Gupta, A.; Narwal, R.; Steele, K.; Gu, Y.; Karakunnel, J.J.; et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016, 17, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Murakami, K.; Inoue, A.; Yonezawa, T.; Matsuki, N. CCR4 Blockade Depletes Regulatory T Cells and Prolongs Survival in a Canine Model of Bladder Cancer. Cancer Immunol. Res. 2019, 7, 1175–1187. [Google Scholar] [CrossRef]
- Leyland, R.; Watkins, A.; Mulgrew, K.A.; Holoweckyj, N.; Bamber, L.; Tigue, N.J.; Offer, E.; Andrews, J.; Yan, L.; Mullins, S.; et al. A Novel Murine GITR Ligand Fusion Protein Induces Antitumor Activity as a Monotherapy That Is Further Enhanced in Combination with an OX40 Agonist. Clin. Cancer Res. 2017, 23, 3416–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumi, K.; Miyakawa, R.; Shibasaki, C.; Henmi, M.; Mizoguchi, Y.; Ueda, R.; Hashimoto, H.; Hiraoka, N.; Yoshida, T.; Aoki, K. Local Administration of GITR Agonistic Antibody Induces a Stronger Antitumor Immunity than Systemic Delivery. Sci. Rep. 2019, 9, 5562. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.; Balkwill, F.R. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef] [Green Version]
- Rauh, M.J.; Sly, L.M.; Kalesnikoff, J.; Hughes, M.R.; Cao, L.P.; Lam, V.; Krystal, G. The role of SHIP1 in macrophage programming and activation. Biochem. Soc. Trans. 2004, 32, 785–788. [Google Scholar] [CrossRef]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Jutras, I.; Desjardins, M. Phagocytosis: At the crossroads of innate and adaptive immunity. Annu. Rev. Cell Dev. Biol. 2005, 21, 511–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Lian, S.; Xie, R.; Ye, Y.; Xie, X.; Li, S.; Lu, Y.; Li, B.; Cheng, Y.; Katanaev, V.L.; Jia, L. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine 2019, 42, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In Vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Idoyaga, J.; Lubkin, A.; Fiorese, C.; Lahoud, M.H.; Caminschi, I.; Huang, Y.; Rodriguez, A.; Clausen, B.E.; Park, C.G.; Trumpfheller, C.; et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl. Acad. Sci. USA 2011, 108, 2384–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, D.; Mourao-Sa, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Invest. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.Z.; Keler, T.; Odunsi, K.; Old, L.J.; Ritter, G.; et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J. Immunol. 2011, 186, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232–251. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.Y.; Chung, W.H.; Chu, M.T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 4325874. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Papachristofilou, A.; Weiss, C.; Fruh, M.; Cathomas, R.; Hilbe, W.; Wehler, T.; Rippin, G.; Koch, S.D.; Scheel, B.; et al. Phase Ib study evaluating a self-adjuvanted mRNA cancer vaccine (RNActive(R)) combined with local radiation as consolidation and maintenance treatment for patients with stage IV non-small cell lung cancer. BMC Cancer 2014, 14, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, M.; Schroder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Strbo, N.; Garcia-Soto, A.; Schreiber, T.H.; Podack, E.R. Secreted heat shock protein gp96-Ig: Next-generation vaccines for cancer and infectious diseases. Immunol. Res. 2013, 57, 311–325. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Park, J.H.; Brentjens, R.J. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov. Med. 2010, 9, 277–288. [Google Scholar]
- Neelapu, S.S.; Locke, F.L.; Go, W.Y. CAR T-Cell Therapy in Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1065. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Song, Y.; Liu, D. Clinical trials of CAR-T cells in China. J. Hematol. Oncol. 2017, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Leighl, N.B.; Wu, Y.L.; Zhong, W.Z. Emerging therapies for non-small cell lung cancer. J. Hematol. Oncol. 2019, 12, 45. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Stimulatory Molecule | Ligand | Expression on Teff | Effects on Teff Function | Expression on Treg | Effects on Treg Function |
|---|---|---|---|---|---|
| CTLA-4 | CD80, CD86 | induced by stimulation | ↓ | constitutively expressed | ↑ |
| PD-1 | PD-L1 | induced by stimulation | ↓ | constitutively expressed | ↑ |
| ICOS | ICOSL | induced by stimulation | ↑ | constitutively expressed | ↑ |
| TIGIT | CD155 | induced by stimulation | ↓ | constitutively expressed | ↑ |
| CD27 | CD70 | constitutively expressed | ↑ | constitutively expressed | ↑ |
| 4-1BB | 4-1BBL | induced by stimulation | ↑ | - | - |
| OX40 | OX40L | induced by stimulation | ↑ | constitutively expressed | ↓ |
| GITR | GITRL | induced by stimulation | ↑ | constitutively expressed | ↓ |
| CD226 | CD155 | constitutively expressed | ↑ | - | - |
| BTLA | HVEM | constitutively expressed | ↓ | - | - |
| TIM-3 | galectin-9 | induced by stimulation | ↓ | constitutively expressed | ↑ |
| LAG-3 | MHC-II | induced by stimulation | ↓ | induced by stimulation | ↑ |
| Clinical Trials, Gov Identifier | Phase | Intervention | Trial Design | Cancer Type | Primary End Points |
|---|---|---|---|---|---|
| Monotherapy Trials | |||||
| NCT03763149 | I | Anti-CD47 antibody (IBI188) | Dose escalation | advanced malignancies and lymphoma | safety and tolerability |
| NCT02678338 | I | Anti-CD47 antibody (Hu5F9-G4) | Dose escalation | hematological malignancies | tolerability |
| NCT02216409 | I | Anti-CD47 antibody (Hu5F9-G4) | Dose escalation | solid tumors | safety and tolerability |
| NCT03834948 | I | Anti-CD47 antibody (AO-176) | Dose escalation Dose expansion | solid tumors | safety and tolerability |
| NCT03013218 | I | high-affinity SIRPα fusion protein (ALX148) | Dose escalation | solid tumors and lymphoma | dose-limiting toxicity |
| NCT03512340 | I | Anti-CD47 antibody (SRF231) | Dose escalation Dose expansion | solid tumors hematological malignancies | safety and tolerability |
| Combination Trials | |||||
| NCT02367196 | I | Anti-CD47 antibody (CC-90002) alone or in combination with rituximab | Dose escalation | solid tumors and hematological malignancies | safety and tolerability |
| NCT02663518 | I | SIRPαFc (TTI-621) alone or in combination with rituximab or nivolumab | Dose escalation | relapsed/refractory hematological and solid malignancies | safety and tolerability |
| NCT02890368 | I | SIRPαFc (TTI-621) alone or in combination with an anti-PD-1/PD-L1 agent, pegylated IFNα2a, T-VEC or radiation | non-randomized parallel assignment | solid tumors and mycosis fungoides | optimal delivery regimen |
| NCT03248479 | Ib | Anti-CD47 antibody (Hu5F9-G4) alone or in combination with azactidine | non-randomized | AML and MDS | safety and tolerability |
| NCT02953509 | Ib/II | Anti-CD47 antibody (Hu5F9-G4) in combination with rituximab | single-arm, non-randomized | refractory/relapsed non-Hodgkin lymphoma | safety and tolerability |
| NCT02953782 | I/II | Anti-CD47 antibody (Hu5F9-G4) in combination with cetuximab | single-arm, non-randomized | solid tumors and CRC | safety and tolerability |
| Tumor Antigens | Proteins | Specificity | Advantages | Disadvantages |
|---|---|---|---|---|
| Differentiation antigens | MART1, GP100, PAP, CEA | Low | high prevalence, cheap off-the-shelf products, allow conjugation | high probability of nonspecificity and side effects |
| Overexpressed antigens | WT1, MUC1, ERBB2 | Low | high prevalence, cheap off-the-shelf products, allow conjugation | high probability of nonspecificity and side effects |
| Viral antigens | HPV-, EBV-derived proteins | High | very specific, allow conjugation | limited prevalence of virus-associated tumors |
| Cancer-Germline antigens | NY-ESO-1, MAGE, GAGE, BAGE | High | specific, cheap off-the-shelf products allow conjugation | not exclusive to cancer |
| Mutated neoantigens | mutated tumor neoantigens | Highest | very specific, allow conjugation | expensive, labor- and technology intensive |
| Whole tumor antigens | lysate of cancer material | Variable | no need for neoantigen identification, contain additional DC-activating factors | limited cancer material, uncontrolled material |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunimasa, K.; Goto, T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. Int. J. Mol. Sci. 2020, 21, 597. https://doi.org/10.3390/ijms21020597
Kunimasa K, Goto T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. International Journal of Molecular Sciences. 2020; 21(2):597. https://doi.org/10.3390/ijms21020597
Chicago/Turabian StyleKunimasa, Kei, and Taichiro Goto. 2020. "Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges" International Journal of Molecular Sciences 21, no. 2: 597. https://doi.org/10.3390/ijms21020597
APA StyleKunimasa, K., & Goto, T. (2020). Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. International Journal of Molecular Sciences, 21(2), 597. https://doi.org/10.3390/ijms21020597