The Cutaneous Wound Innate Immunological Microenvironment


Abstract
:1. Wounding Induces an Immunological Disruption to the Skin Barrier
2. Cutaneous Wounds as Entry Site for Infectious Agents
3. Innate Immune Response to Acute Cutaneous Wounds: A Brief Overview
4. Sensing a Wound: Immune Microenvironment Is Dependent on Neuroimmune Signaling
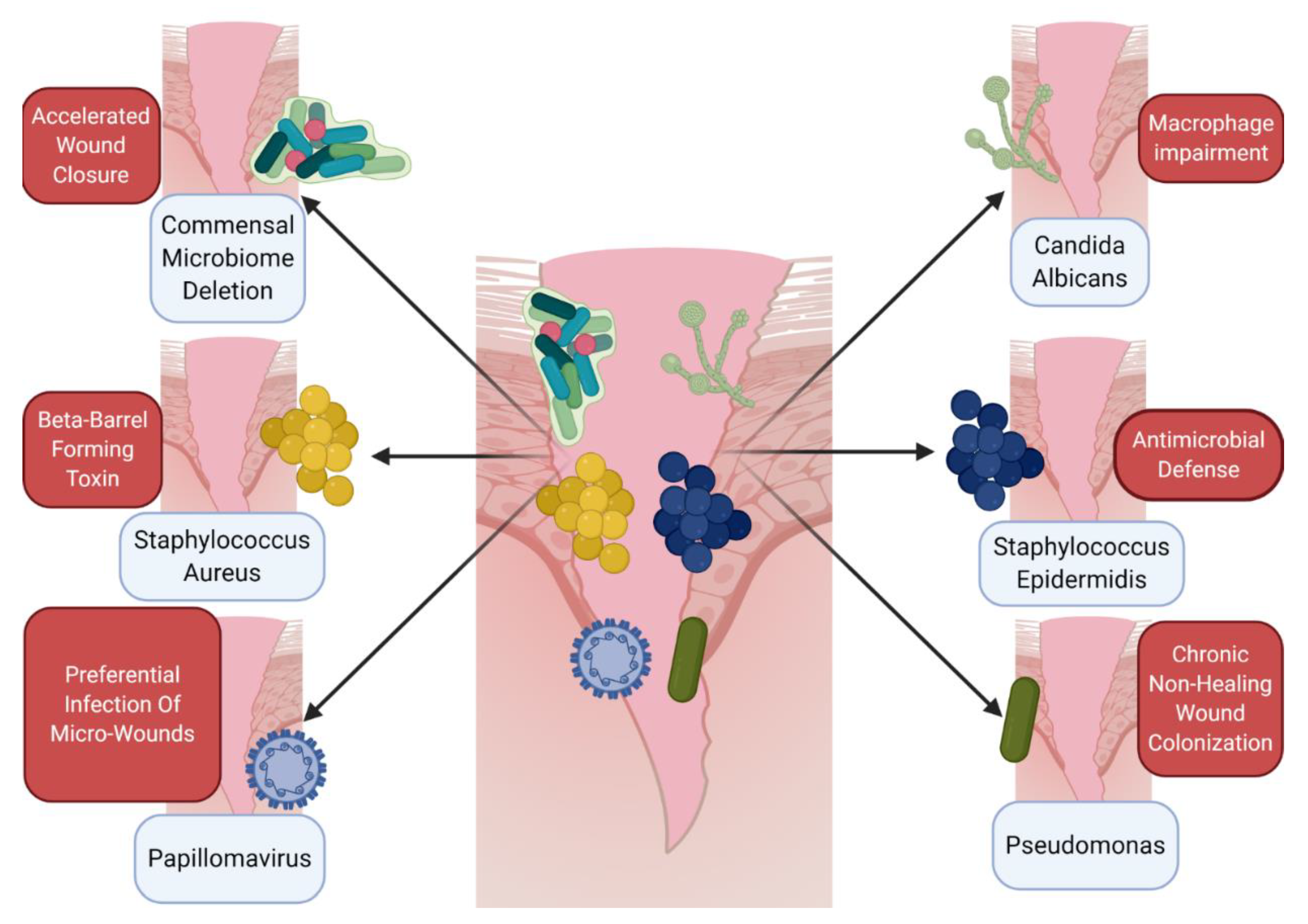
5. Host–Microbe Interactions in the Wound as a Component of the Immune Microenvironment
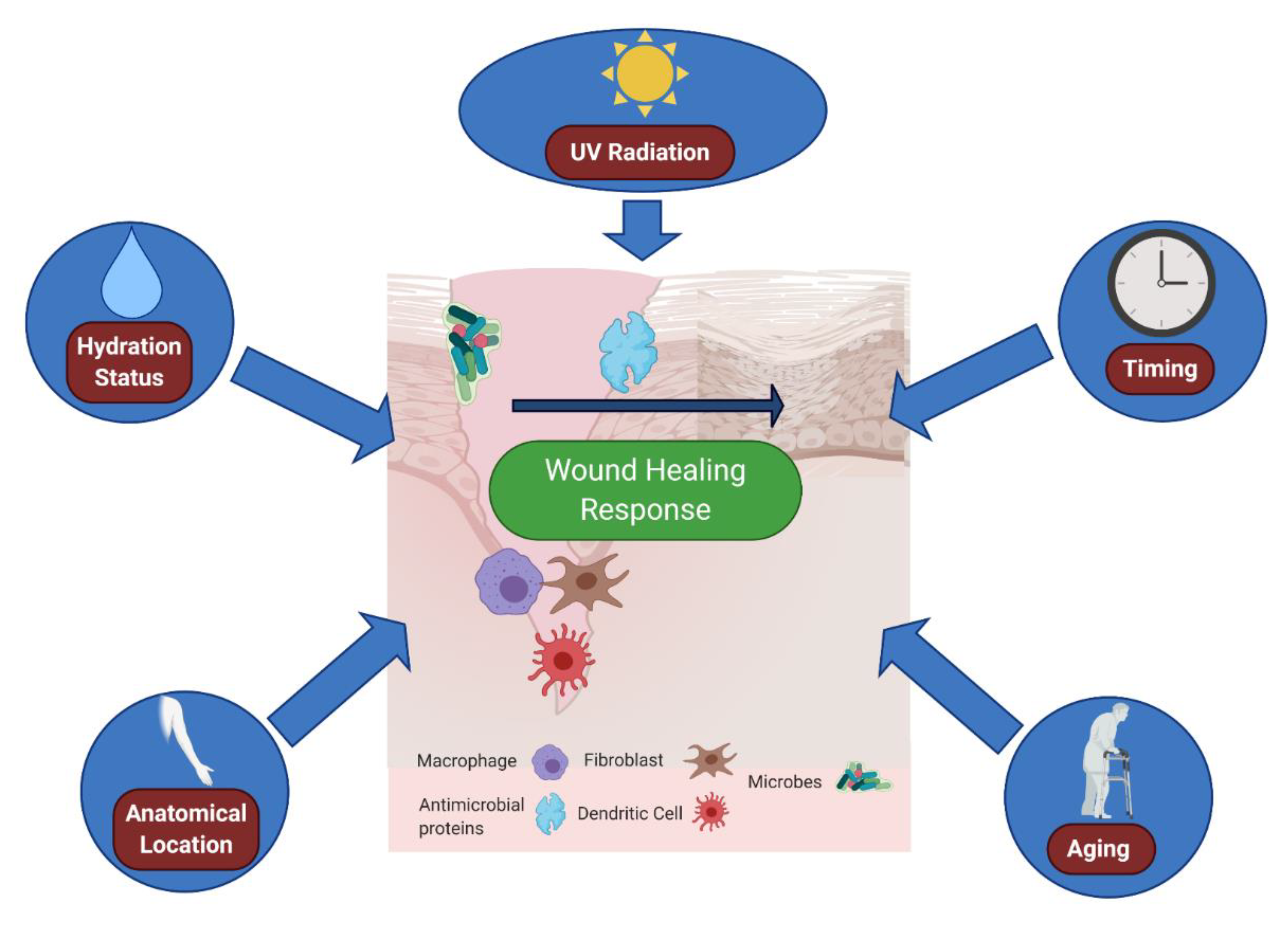
6. Environmental Effects: The Outside World Alters the Cutaneous Wound Environment

{kind=link}
{kind=link}
| Microenvironment Component | Outcome(s) | Reference(s) | |
|---|---|---|---|
| Internal | Neural Sensation | Denervated skin heals at slower rates; | [42] |
| TRPV1 nerve fibers activate host immune defenses | [44,45] | ||
| Internal | Wound Location | Immune cell numbers vary with body site | [80] |
| Internal | Age | Elderly Skin heals slower than younger skin; | [81] |
| Inflammation/repair spectrum is impaired in aged skin | [82] | ||
| External | Cutaneous Bacteria | Microbiome deletion potentiates wound closure; | [54] |
| Commensal microbes can promote antimicrobial defense; | [57] | ||
| Microbiome is altered in chronic, non-healing wounds | [64,65] | ||
| External | Cutaneous Fungus | Cutaneous fungal communities are predictive of wound healing time | [67] |
| External | Cutaneous Virus | IL-27 promotes antiviral defense and healing in cutaneous wounds | [47] |
| External | Moisture | Emollients can promote antibacterial defenses; | [76] |
| Skin moisture levels directly impact wound healing rate | [75] | ||
| External | UV Radiation | UVB radiation activates Type I interferon responses; | [83] |
| UVB radiation can directly stimulate wound healing | [84] | ||
| External | Time of Wound | Fibroblast migration and wound healing varies with time of wound | [85] |

7. An Aging Microenvironment
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallo, R.L. Human Skin Is the Largest Epithelial Surface for Interaction with Microbes. J. Investig. Dermatol. 2017, 137, 1213–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyerich, S.; Eyerich, K.; Traidl-Hoffmann, C.; Biedermann, T. Cutaneous Barriers and Skin Immunity: Differentiating A Connected Network. Trends Immunol. 2018, 39, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.W.; Watkins, K.; Hewlett, A. Infection control through the ages. Am. J. Infect. Control. 2012, 40, 35–42. [Google Scholar] [CrossRef]
- Haagensen, C.D.; Lloyd, W.E.B. A Hundred Years of Medicine; Beard Books: Frederick, MD, USA, 1943. [Google Scholar]
- Mayon-White, R.T.; Ducel, G.; Kereselidze, T.; Tikomirov, E. An international survey of the prevalence of hospital-acquired infection. J. Hosp. Infect. 1988, 11, 43–48. [Google Scholar] [CrossRef]
- Garner, B.H.; Anderson, D.J. Surgical Site Infections: An Update. Infect. Dis. Clin. N. Am. 2016, 30, 909–929. [Google Scholar] [CrossRef]
- McKibben, L.; Horan, T.; Tokars, J.I.; Fowler, G.; Cardo, D.M.; Pearson, M.L.; Brennan, P.J.; The Healthcare Infection Control Practices Advisory Committee. Guidance on Public Reporting of Healthcare-Associated Infections: Recommendations of the Healthcare Infection Control Practices Advisory Committee. Am. J. Infect. Control. 2005, 33, 217–226. [Google Scholar] [CrossRef]
- Awad, S.S. Adherence to surgical care improvement project measures and post-operative surgical site infections. Surg. Infect. 2012, 13, 234–237. [Google Scholar] [CrossRef]
- Zimlichman, E.; Henderson, D.; Tamir, O.; Franz, C.; Song, P.; Yamin, C.K.; Keohane, C.; Denham, C.R.; Bates, D.W. Health care-associated infections: A meta-analysis of costs and financial impact on the US health care system. JAMA Intern. Med. 2013, 173, 2039–2046. [Google Scholar] [CrossRef]
- Sen, C.K. Human Wounds and Its Burden: An Updated Compendium of Estimates. Adv. Wound Care 2019, 8, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Makrantonaki, E.; Wlaschek, M.; Scharffetter-Kochanek, K. Pathogenesis of wound healing disorders in the elderly. JDDG J. Der Dtsch. Dermatol. Ges. 2017, 15, 255–275. [Google Scholar] [CrossRef]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The diabetes pandemic and associated infections: Suggestions for clinical microbiology. Rev. Med. Microbiol. 2019, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bowler, P.G.; Duerden, B.I.; Armstrong, D.G. Wound microbiology and associated approaches to wound management. Clin. Microbiol. Rev. 2001, 14, 244–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, M.G.; Holzer, K.J.; Carbone, J.T.; Salas-Wright, C.P. Arthropod Bites and Stings Treated in Emergency Departments: Recent Trends and Correlates. Wilderness Environ. Med. 2019, 30, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.; McDowell, R.H. Insect Bites. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Vasievich, M.P.; Villarreal, J.D.M.; Tomecki, K.J. Got the Travel Bug? A Review of Common Infections, Infestations, Bites, and Stings Among Returning Travelers. Am. J. Clin. Dermatol. 2016, 17, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Brito, A.F.; Swetnam, D.M.; Vogels, C.B.F.; Tokarz, R.E.; Andersen, K.G.; Smith, R.C.; Bedford, T.; Grubaugh, N.D. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. 2019, 15, e1008042. [Google Scholar] [CrossRef] [Green Version]
- Bos, S.; Gadea, G.; Despres, P. Dengue: A growing threat requiring vaccine development for disease prevention. Pathog. Glob. Health 2018, 112, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Hills, S.L.; Fischer, M.; Petersen, L.R. Epidemiology of Zika Virus Infection. J. Infect. Dis. 2017, 216, S868–S874. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound Care 2016, 5, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Sub-Cell. Biochem. 2017, 82, 405–456. [Google Scholar]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Ali, R.A.; Wuescher, L.M.; Worth, R.G. Platelets: Essential components of the immune system. Curr. Trends Immunol. 2015, 16, 65–78. [Google Scholar]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [Green Version]
- Munir, S.; Basu, A.; Maity, P.; Krug, L.; Haas, P.; Jiang, D.; Strauss, G.; Wlaschek, M.; Geiger, H.; Singh, K.; et al. TLR4-dependent shaping of the wound site by MSCs accelerates wound healing. EMBO Rep. 2020, 21, e48777. [Google Scholar] [CrossRef]
- Büchau, A.S.; Hassan, M.; Kukova, G.; Lewerenz, V.; Kellermann, S.; Würthner, J.U.; Wolf, R.; Walz, M.; Gallo, R.L.; Ruzicka, T. S100A15, an Antimicrobial Protein of the Skin: Regulation by E. coli through Toll-Like Receptor 4. J. Investig. Dermatol. 2007, 127, 2596–2604. [Google Scholar]
- Bdeir, K.; Gollomp, K.; Stasiak, M.; Mei, J.; Papiewska-Pajak, I.; Zhao, G.; Worthen, G.S.; Cines, D.B.; Poncz, M.; Kowalska, M.A. Platelet-Specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2016, 56, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low concentrations of neutrophil extracellular traps induce proliferation in human keratinocytes via NF-kB activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef]
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; DeMarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Paige, J.T.; Kremer, M.; Landry, J.; Hatfield, S.A.; Wathieu, D.; Brug, A.; Lightell, D.J.; Spiller, K.L.; Woods, T.C. Modulation of inflammation in wounds of diabetic patients treated with porcine urinary bladder matrix. Regen. Med. 2019, 14, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, M.; Baguneid, M.; Bayat, A. The Role of Neuromediators and Innervation in Cutaneous Wound Healing. Acta Derm. Venereol. 2016, 96, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal Control of Skin Function: The Skin as a Neuroimmunoendocrine Organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef]
- Fukai, T.; Takeda, A.; Uchinuma, E. Wound healing in denervated rat skin. Wound Repair Regen. 2005, 13, 175–180. [Google Scholar] [CrossRef]
- Souza, B.R.; Cardoso, J.F.; Amadeu, T.P.; Desmoulière, A.; Costa, A.M.A. Sympathetic denervation accelerates wound contraction but delays reepithelialization in rats. Wound Repair Regen. 2005, 13, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.A.; Edwards, T.N.; Liu, A.W.; Hirai, T.; Jones, M.R.; Wu, J.; Li, Y.; Zhang, S.; Ho, J.; Davis, B.M.; et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019, 178, 919–932.e14. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.S.; Hemmers, S.; Garijo, O.; Chabod, M.; Mowen, K.; Witherden, D.A.; Havran, W.L. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Investig. 2013, 123, 4364–4374. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Suwanpradid, J.; Sanchez-Lagunes, R.; Choi, H.W.; Hoang, P.; Wang, D.; Abraham, S.N.; Macleod, A.S. IL-27 Facilitates Skin Wound Healing through Induction of Epidermal Proliferation and Host Defense. J. Investig. Dermatol. 2017, 137, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Shook, B.; Xiao, E.; Kumamoto, Y.; Iwasaki, A.; Horsley, V. CD301b+ Macrophages Are Essential for Effective Skin Wound Healing. J. Investig. Dermatol. 2016, 136, 1885–1891. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Kumamoto, Y.; Gopinath, S.; Iwasaki, A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat. Commun. 2016, 7, 13346. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Johnson, T.R.; Gómez, B.I.; McIntyre, M.K.; Dubick, M.A.; Christy, R.J.; Nicholson, S.E.; Burmeister, D.M. The Cutaneous Microbiome and Wounds: New Molecular Targets to Promote Wound Healing. Int. J. Mol. Sci. 2018, 19, 2699. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, M.; Vicaretti, M.; Sparks, J.; Bansal, S.; Bush, S.; Liu, M.; Darling, A.; Harry, E.; Burke, C.M. A longitudinal study of the diabetic skin and wound microbiome. PeerJ 2017, 5, e3543. [Google Scholar] [CrossRef]
- Bartow-McKenney, C.; Hannigan, G.D.; Horwinski, J.; Hesketh, P.; Horan, A.D.; Mehta, S.; Grice, E.A. The microbiota of traumatic, open fracture wounds is associated with mechanism of injury. Wound Repair Regen. 2018, 26, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Canesso, M.C.C.; Vieira, A.T.; Castro, T.B.R.; Schirmer, B.G.A.; Cisalpino, D.; Martins, F.S.; Rachid, M.A.; Nicoli, J.R.; Teixeira, M.M.; Barcelos, L.S. Skin Wound Healing Is Accelerated and Scarless in the Absence of Commensal Microbiota. J. Immunol. 2014, 193, 5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linehan, J.L.; Harrison, O.J.; Han, S.J.; Byrd, A.L.; Vujkovic-Cvijin, I.; Villarino, A.V.; Sen, S.K.; Shaik, J.; Smelkinson, M.; Tamoutounour, S.; et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 2018, 172, 784–796.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.-R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate Toll-like receptor 3–dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective Antimicrobial Action Is Provided by Phenol-Soluble Modulins Derived from Staphylococcus epidermidis, a Normal Resident of the Skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol 2015, 13, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Uwamahoro, N.; Verma-Gaur, J.; Shen, H.-H.; Qu, Y.; Lewis, R.; Lu, J.; Bambery, K.; Masters, S.L.; Vince, J.E.; Naderer, T.; et al. The Pathogen Candida albicans Hijacks Pyroptosis for Escape from Macrophages. mBio 2014, 5, e00003-14. [Google Scholar] [CrossRef] [Green Version]
- Hufbauer, M.; Akgül, B. Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis. Viruses 2017, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.; Crompton, R.A.; Thomason, H.A.; Campbell, L.; Singh, G.; McBain, A.J.; Cruickshank, S.M.; Hardman, M.J. Cutaneous Nod2 Expression Regulates the Skin Microbiome and Wound Healing in a Murine Model. J. Investig. Dermatol. 2017, 137, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.; Campbell, L.; Crompton, R.A.; Singh, G.; McHugh, B.J.; Davidson, D.J.; McBain, A.J.; Cruickshank, S.M.; Hardman, M.J. Microbial Host Interactions and Impaired Wound Healing in Mice and Humans: Defining a Role for BD14 and NOD2. J. Investig. Dermatol. 2018, 138, 2264–2274. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.; Williams, H.; Crompton, R.A.; Cruickshank, S.M.; Hardman, M.J. Nod2 deficiency impairs inflammatory and epithelial aspects of the cutaneous wound-healing response. J. Pathol. 2013, 229, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Hanson, J.D.; Rees, E.J.; Koenig, L.D.; Phillips, C.D.; Wolcott, R.A.; Cox, S.B.; White, J.S. Analysis of the chronic wound microbiota of 2963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 2016, 24, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Verbanic, S.; Shen, Y.; Lee, J.; Deacon, J.M.; Chen, I.A. Microbial predictors of healing and short-term effect of debridement on the microbiome of chronic wounds. NPJ Biofilms Microbiomes 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Ruffin, M.; Brochiero, E. Repair Process Impairment by Pseudomonas aeruginosa in Epithelial Tissues: Major Features and Potential Therapeutic Avenues. Front. Cell. Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalan, L.; Loesche, M.; Hodkinson, B.P.; Heilmann, K.; Ruthel, G.; Gardner, S.E.; Grice, E.A. Redefining the Chronic-Wound Microbiome: Fungal Communities Are Prevalent, Dynamic, and Associated with Delayed Healing. mBio 2016, 7, e01058-16. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The Human Skin Double-Stranded DNA Virome: Topographical and Temporal Diversity, Genetic Enrichment, and Dynamic Associations with the Host Microbiome. mBio 2015, 6, e01578-15. [Google Scholar] [CrossRef] [Green Version]
- Foulongne, V.; Sauvage, V.; Hebert, C.; Dereure, O.; Cheval, J.; Gouilh, M.A.; Pariente, K.; Segondy, M.; Burguière, A.; Manuguerra, J.C.; et al. Human skin microbiota: High diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS ONE 2012, 7, e38499. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.A.; Kastelein, R. Interleukin-27: Balancing protective and pathological immunity. Immunity 2012, 37, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Kwock, J.T.; Handfield, C.; Suwanpradid, J.; Hoang, P.; McFadden, M.J.; Labagnara, K.F.; Floyd, L.; Shannon, J.; Uppala, R.; Sarkar, M.K.; et al. IL-27 signaling activates skin cells to induce innate antiviral proteins and protects against Zika virus infection. Sci. Adv. 2020, 6, eaay3245. [Google Scholar] [CrossRef] [Green Version]
- Marks, R. The stratum corneum barrier: The final frontier. J. Nutr. 2004, 134, 2017s–2021s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparr, E.; Millecamps, D.; Isoir, M.; Burnier, V.; Larsson, Å.; Cabane, B. Controlling the hydration of the skin though the application of occluding barrier creams. J. R. Soc. Interface 2012, 10, 20120788. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Xia, H.; He, W.; Li, Z.; Zhao, J.; Liu, B.; Wang, Y.; Lei, Q.; Kong, Y.; Bai, Y.; et al. Controlled water vapor transmission rate promotes wound-healing via wound re-epithelialization and contraction enhancement. Sci. Rep. 2016, 6, 24596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnowicki, T.; Malajian, D.; Khattri, S.; Correa da Rosa, J.; Dutt, R.; Finney, R.; Dhingra, N.; Xiangyu, P.; Xu, H.; Estrada, Y.D.; et al. Petrolatum: Barrier repair and antimicrobial responses underlying this “inert” moisturizer. J. Allergy Clin. Immunol. 2016, 137, 1091–1102.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Q.; Ku, A.T.; Nishino, Y.; Howard, J.M.; Rao, A.S.; Shaver, T.M.; Garcia, G.E.; Le, D.N.; Karlin, K.L.; Westbrook, T.F.; et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat. Commun. 2014, 5, 4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Investig. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Field, C.K.; Kerstein, M.D. Overview of wound healing in a moist environment. Am. J. Surg. 1994, 167, S2–S6. [Google Scholar] [CrossRef]
- Omine, Y.; Hinata, N.; Yamamoto, M.; Kasahara, M.; Matsunaga, S.; Murakami, G.; Abe, S.-I. Regional differences in the density of Langerhans cells, CD8-positive T lymphocytes and CD68-positive macrophages: A preliminary study using elderly donated cadavers. Anat. Cell Biol. 2015, 48, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, G.S.; Mills, S.J.; Ashworth, J.J. Ageing and wound healing. Biogerontology 2002, 3, 337–345. [Google Scholar] [CrossRef]
- Haustead, D.J.; Stevenson, A.; Saxena, V.; Marriage, F.; Firth, M.; Silla, R.; Martin, L.; Adcroft, K.F.; Rea, S.; Day, P.J.; et al. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB. Sci. Rep. 2016, 6, 26846. [Google Scholar] [CrossRef] [Green Version]
- Skopelja-Gardner, S.; An, J.; Tai, J.; Tanaka, L.; Sun, X.; Hermanson, P.; Baum, R.; Kawasumi, M.; Green, R.; Gale, M., Jr.; et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci. Rep. 2020, 10, 7908. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Avci, P.; Dai, T.; Huang, Y.-Y.; Hamblin, M.R. Ultraviolet Radiation in Wound Care: Sterilization and Stimulation. Adv. Wound Care 2013, 2, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Hoyle, N.P.; Seinkmane, E.; Putker, M.; Feeney, K.A.; Krogager, T.P.; Chesham, J.E.; Bray, L.K.; Thomas, J.M.; Dunn, K.; Blaikley, J.; et al. Circadian actin dynamics drive rhythmic fibroblast mobilization during wound healing. Sci. Transl. Med. 2017, 9, eaal2774. [Google Scholar] [CrossRef] [Green Version]
- Pick, R.; He, W.; Chen, C.-S.; Scheiermann, C. Time-of-Day-Dependent Trafficking and Function of Leukocyte Subsets. Trends Immunol. 2019, 40, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced Neutrophil Chemotaxis and Infiltration Contributes to Delayed Resolution of Cutaneous Wound Infection with Advanced Age. J. Immunol. 2013, 190, 1746. [Google Scholar] [CrossRef] [Green Version]
- Kopcewicz, M.; Walendzik, K.; Bukowska, J.; Kur-Piotrowska, A.; Machcinska, S.; Gimble, J.M.; Gawronska-Kozak, B. Cutaneous wound healing in aged, high fat diet-induced obese female or male C57BL/6 mice. Aging 2020, 12, 7066–7111. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Garcia, B.; Murray, C.K.; Vrahas, M.S.; Hamblin, M.R. UVC Light Prophylaxis for Cutaneous Wound Infections in Mice. Antimicrob. Agents Chemother. 2012, 56, 3841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, J.; Meller, S.; Conrad, C.; Di Nardo, A.; Homey, B.; Lauerma, A.; Arai, N.; Gallo, R.L.; Digiovanni, J.; Gilliet, M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med. 2010, 207, 2921–2930. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yue, J.; Lei, Q.; Gou, X.; Chen, S.-Y.; He, Y.-Y.; Wu, X. Ultraviolet B Inhibits Skin Wound Healing by Affecting Focal Adhesion Dynamics. Photochem. Photobiol. 2015, 91, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Azzouz, D.; Khan, M.A.; Sweezey, N.; Palaniyar, N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Patra, V.; Wagner, K.; Arulampalam, V.; Wolf, P. Skin Microbiome Modulates the Effect of Ultraviolet Radiation on Cellular Response and Immune Function. iScience 2019, 15, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paatela, E.; Munson, D.; Kikyo, N. Circadian Regulation in Tissue Regeneration. Int. J. Mol. Sci. 2019, 20, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrecque, N.; Cermakian, N. Circadian Clocks in the Immune System. J. Biol. Rhythm. 2015, 30, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.K.; Liou, Y.-H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.-S.; Lee, C.-H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9, e54090. [Google Scholar] [CrossRef]
- Greenberg, E.N.; Marshall, M.E.; Jin, S.; Venkatesh, S.; Dragan, M.; Tsoi, L.C.; Gudjonsson, J.E.; Nie, Q.; Takahashi, J.S.; Andersen, B. Circadian control of interferon-sensitive gene expression in murine skin. Proc. Natl. Acad. Sci. USA 2020, 117, 5761–5771. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.-D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407. [Google Scholar] [CrossRef] [Green Version]
- Chopra, K.; Calva, D.; Sosin, M.; Tadisina, K.K.; Banda, A.; De La Cruz, C.; Chaudhry, M.R.; Legesse, T.; Drachenberg, C.B.; Manson, P.N.; et al. A Comprehensive Examination of Topographic Thickness of Skin in the Human Face. Aesthetic Surg. J. 2015, 35, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Rittié, L. Cellular mechanisms of skin repair in humans and other mammals. J. Cell Commun. Signal. 2016, 10, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Sinha, M.; Sen, C.K.; Singh, K.; Das, A.; Ghatak, S.; Rhea, B.; Blackstone, B.; Powell, H.M.; Khanna, S.; Roy, S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018, 9, 936. [Google Scholar] [CrossRef] [Green Version]
- Deckers, J.; Hammad, H.; Hoste, E. Langerhans Cells: Sensing the Environment in Health and Disease. Front. Immunol. 2018, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Tur, E.; Tur, M.; Maibach, H.I.; Guy, R.H. Basal perfusion of the cutaneous microcirculation: Measurements as a function of anatomic position. J. Investig. Dermatol. 1983, 81, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaglstein, W.H. Wound healing and aging. Dermatol. Clin. 1986, 4, 481–484. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruber, J. Age-Related Aspects of Cutaneous Wound Healing: A Mini-Review. Gerontology 2013, 59, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab. Investig. 1998, 78, 47–58. [Google Scholar]
- Thornton, M.J. Estrogens and aging skin. Dermatoendocrinology 2013, 5, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Dodsworth, J.; van Boxtel, E.; Tarnuzzer, R.W.; Horan, M.A.; Schultz, G.S.; Ferguson, M.W. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat. Med. 1997, 3, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Liu, S.; Asare, A.; Naik, S.; Levorse, J.; Polak, L.; Lu, C.P.; Nikolova, M.; Pasolli, H.A.; Fuchs, E. Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 2016, 167, 1323–1338.e14. [Google Scholar] [CrossRef] [Green Version]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.-P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, G.T.; Suaya, J.A.; Baxter, R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a U.S. population: A retrospective population-based study. BMC Infect. Dis. 2013, 13, 252. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Je, Y.J.; Kim, C.D.; Lee, Y.H.; Seo, Y.J.; Lee, J.H.; Lee, Y. Can Platelet-rich Plasma Be Used for Skin Rejuvenation? Evaluation of Effects of Platelet-rich Plasma on Human Dermal Fibroblast. Ann. Dermatol. 2011, 23, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Balduini, C.L.; Noris, P. Platelet count and aging. Haematologica 2014, 99, 953–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchner, S.; Lei, V.; MacLeod, A.S. The Cutaneous Wound Innate Immunological Microenvironment. Int. J. Mol. Sci. 2020, 21, 8748. https://doi.org/10.3390/ijms21228748
Kirchner S, Lei V, MacLeod AS. The Cutaneous Wound Innate Immunological Microenvironment. International Journal of Molecular Sciences. 2020; 21(22):8748. https://doi.org/10.3390/ijms21228748
Chicago/Turabian StyleKirchner, Stephen, Vivian Lei, and Amanda S. MacLeod. 2020. "The Cutaneous Wound Innate Immunological Microenvironment" International Journal of Molecular Sciences 21, no. 22: 8748. https://doi.org/10.3390/ijms21228748
APA StyleKirchner, S., Lei, V., & MacLeod, A. S. (2020). The Cutaneous Wound Innate Immunological Microenvironment. International Journal of Molecular Sciences, 21(22), 8748. https://doi.org/10.3390/ijms21228748