The Immune Functions of Keratinocytes in Skin Wound Healing


Abstract
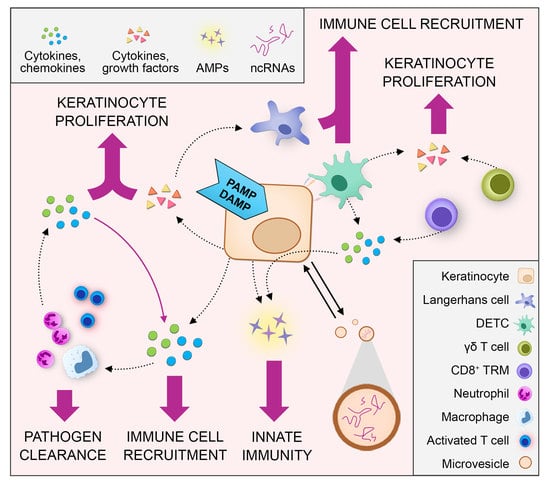
:
1. Introduction
2. Crosstalk between Keratinocytes and Immune Cells during Skin Wound Healing
2.1. Keratinocyte‒Immune Cell Communication via Cytokines and Chemokines
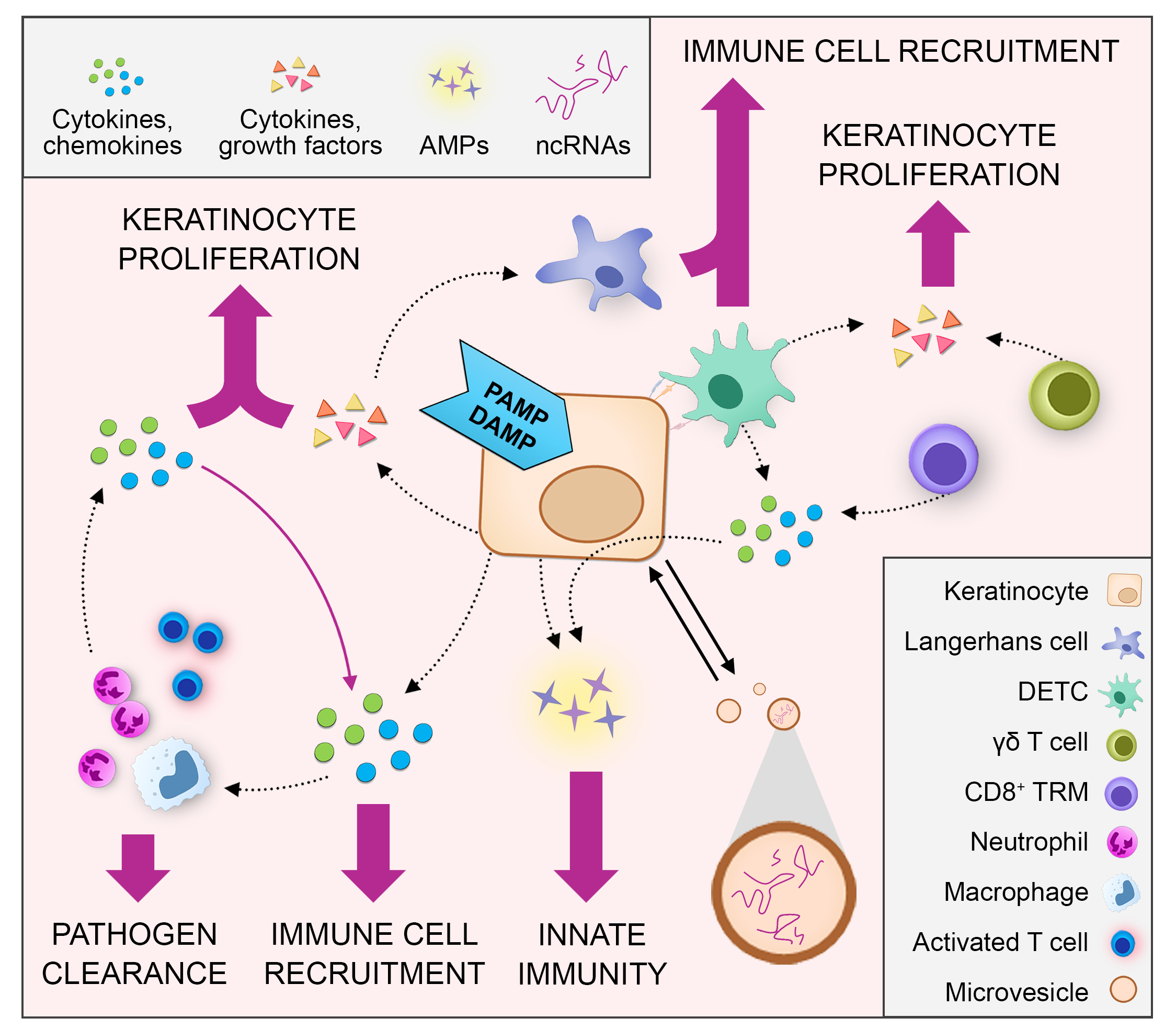
2.1.1. Pattern Recognition Receptor-Mediated Cytokine and Chemokine Expression in Keratinocytes
2.1.2. Other Receptors Expressed by Keratinocytes to Receive Signals from Immune Cells
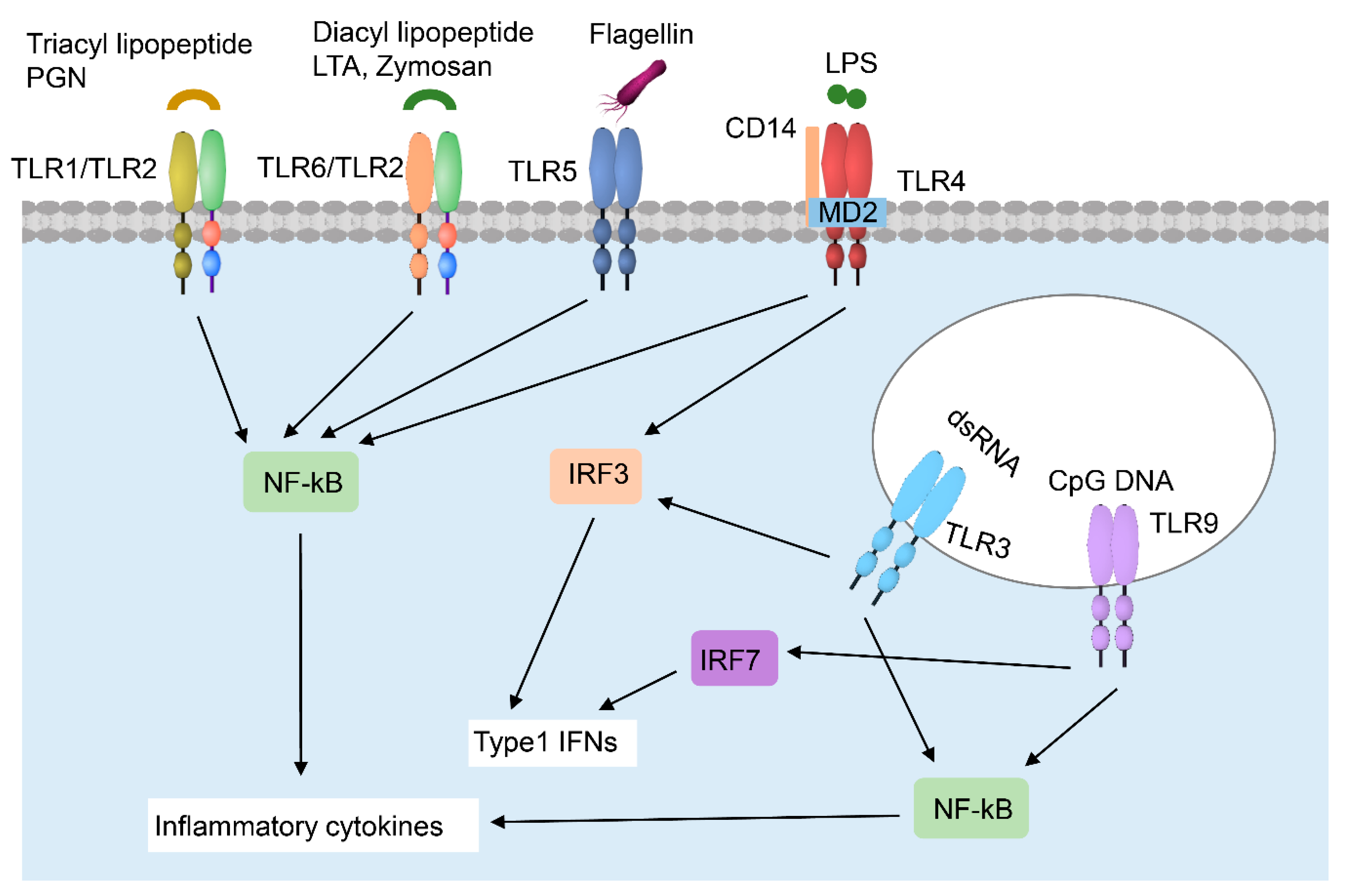
2.1.3. Keratinocyte–Immune Cell Crosstalk in Skin Wound Healing
Neutrophils
Macrophages
Langerhans Cells (LC)
CD8+ Tissue-Resident Memory T (TRM) Cells
Dendritic Epidermal T Cells (DETCs)
2.2. Keratinocyte Crosstalk with Immune Cells via Extracellular Vesicles
2.3. Keratinocyte Interaction with T Cells via Antigen Presentation
3. The Interplay between Keratinocytes and Microorganisms in Skin Wound Healing
3.1. The Role of Microbiota in Wound Healing
3.2. AMPs and Wound Healing
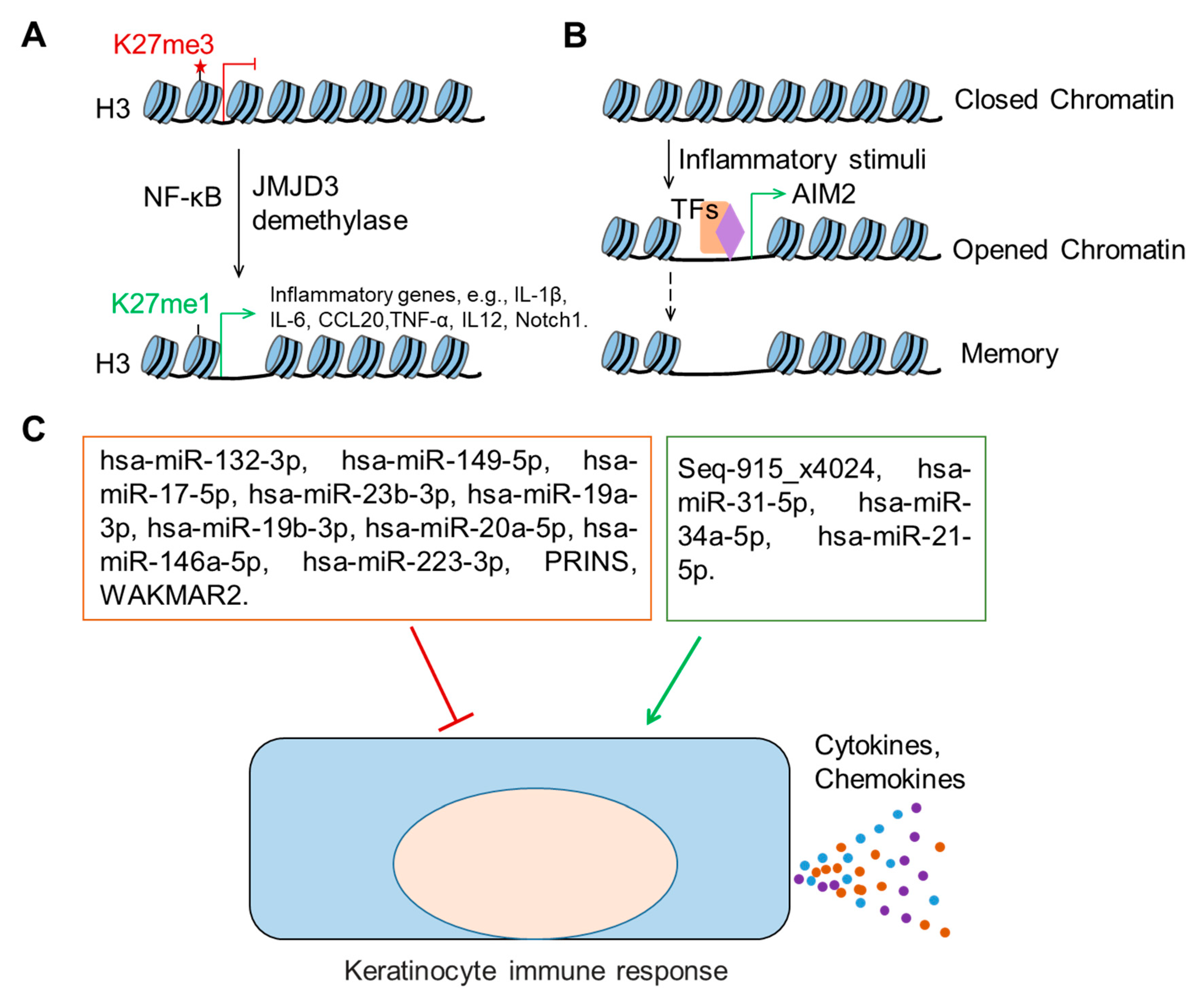
4. Epigenetic Regulation of Keratinocyte Immune Functions in Wound Healing
4.1. Histone Modifications
4.2. Trained Immunity of Epithelial Stem Cells
4.3. Non-Protein-Coding RNAs
4.3.1. microRNAs
4.3.2. lncRNAs
5. Impaired Immune Functions of Keratinocytes in Chronic Nonhealing Wounds
5.1. Cytokines
5.2. TLRs
5.3. AMPs
5.4. MicroRNAs
6. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial peptide |
| APC | Antigen-presenting cell |
| AU | Arterial ulcer |
| DETC | Dendritic epidermal T cell |
| DFU | Diabetic foot ulcer |
| dsRNA | Double-stranded RNA |
| EpSC | Epithelial stem cell |
| EV | Extracellular vesicle |
| ISC | Intestinal stem cell |
| LC | Langerhans cell |
| lncRNA | Long noncoding RNA |
| LPS | Lipopolysaccharide |
| LTA | Lipoteichoic acid |
| MSC | Mesenchymal stem cell |
| miRNA | Micro-RNA |
| ncRNA | Noncoding RNA |
| NET | Neutrophil extracellular trap |
| PAMPs | Pathogen-associated molecular patterns |
| PGN | Peptidoglycan |
| PRR | Pattern-recognition receptor |
| PU | Pressure ulcer |
| RISC | RNA-induced silencing complex |
| ROS | Reactive oxygen species |
| SNP | Single-nucleotide polymorphism |
| ssRNA | Single-stranded RNA |
| TI | Trained immunity |
| TLR | Toll-like receptor |
| UTR | Untranslated region |
| VU | Venous ulcer |
References
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souren, J.M.; Ponec, M.; van Wijk, R. Contraction of collagen by human fibroblasts and keratinocytes. In Vitro Cell. Dev. Biol. 1989, 25, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Isaac, C.; Paggiaro, A.O.; Aldunate, J.L.C.B.; Herson, M.R.; Altran, S.C.; Mônica Beatriz, M.; Ferreira, M.C. Role of keratinocytes in wound contraction: An impact assessment using a model of collagen matrix populated with fibroblasts. Rev. Bras. Cir. Plást. 2011, 26, 402–406. [Google Scholar] [CrossRef]
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nature 2020, 583, 296–302. [Google Scholar] [CrossRef]
- Roupe, K.M.; Nybo, M.; Sjobring, U.; Alberius, P.; Schmidtchen, A.; Sorensen, O.E. Injury is a major inducer of epidermal innate immune responses during wound healing. J. Investig. Derm. 2010, 130, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin microbiota-host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef]
- Lebre, M.C.; van der Aar, A.M.G.; van Baarsen, L.; van Capel, T.M.M.; Schuitemaker, J.H.N.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J. Investig. Derm. 2007, 127, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Miller, L.S.; Modlin, R.L. Toll-like receptors in the skin. Semin. Immunopathol. 2007, 29, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Niebuhr, M.; Baumert, K.; Werfel, T. TLR-2-mediated cytokine and chemokine secretion in human keratinocytes. Exp. Derm. 2010, 19, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Mempel, M.; Voelcker, V.; Köllisch, G.; Plank, C.; Rad, R.; Gerhard, M.; Schnopp, C.; Fraunberger, P.; Walli, A.K.; Ring, J.; et al. Toll-like receptor expression in human keratinocytes: Nuclear factor kappaB controlled gene activation by Staphylococcus aureus is toll-like receptor 2 but not toll-like receptor 4 or platelet activating factor receptor dependent. J. Investig. Derm. 2003, 121, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köllisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Lebre, M.C.; Antons, J.C.; Kalinski, P.; Schuitemaker, J.H.N.; van Capel, T.M.M.; Kapsenberg, M.L.; De Jong, E.C. Double-stranded RNA-exposed human keratinocytes promote Th1 responses by inducing a Type-1 polarized phenotype in dendritic cells: Role of keratinocyte-derived tumor necrosis factor alpha, type I interferons, and interleukin-18. J. Investig. Derm. 2003, 120, 990–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wu, F.; Wu, Z.; Li, Y.; Zhang, S.; Yu, N. IL-17A synergistically enhances TLR3-mediated IL-36γ production by keratinocytes: A potential role in injury-amplified psoriatic inflammation. Exp. Derm. 2019, 28, 233–239. [Google Scholar] [CrossRef]
- Lai, Y.P.; Di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef]
- Li, Z.J.; Sohn, K.-C.; Choi, D.-K.; Shi, G.; Hong, D.; Lee, H.-E.; Whang, K.U.; Lee, Y.H.; Im, M.; Lee, Y.; et al. Roles of TLR7 in activation of NF-κB signaling of keratinocytes by imiquimod. PLoS ONE 2013, 8, e77159. [Google Scholar] [CrossRef] [Green Version]
- Kalali, B.N.; Köllisch, G.; Mages, J.; Müller, T.; Bauer, S.; Wagner, H.; Ring, J.; Lang, R.; Mempel, M.; Ollert, M. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J. Immunol. 2008, 181, 2694–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Y.; Takahashi, T.; Curk, T.; Dobnikar, J.; Gallo, R.L.; Wong, G.C.L. Crystallinity of Double-Stranded RNA-Antimicrobial Peptide Complexes Modulates Toll-Like Receptor 3-Mediated Inflammation. ACS Nano 2017, 11, 12145–12155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-J.; Sen, G.L.; Ward, N.L.; Johnston, A.; Chun, K.; Chen, Y.; Adase, C.; Sanford, J.A.; Gao, N.; Chensee, M.; et al. Antimicrobial Peptide LL37 and MAVS Signaling Drive Interferon-β Production by Epidermal Keratinocytes during Skin Injury. Immunity 2016, 45, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, A.I.; Morizane, S.; Takiguchi, T.; Yamamoto, T.; Yamada, M.; Iwatsuki, K. Interferon-Gamma Enhances TLR3 Expression and Anti-Viral Activity in Keratinocytes. J. Investig. Derm. 2015, 135, 2005–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prens, E.P.; Kant, M.; van Dijk, G.; van der Wel, L.I.; Mourits, S.; van der Fits, L. IFN-alpha enhances poly-IC responses in human keratinocytes by inducing expression of cytosolic innate RNA receptors: Relevance for psoriasis. J. Investig. Derm. 2008, 128, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Borkowski, A.W.; Park, K.; Uchida, Y.; Gallo, R.L. Activation of TLR3 in keratinocytes increases expression of genes involved in formation of the epidermis, lipid accumulation, and epidermal organelles. J. Investig. Derm. 2013, 133, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-Guided Phenotypic Switch of M1 to M2 Macrophages for Cutaneous Wound Healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Chen, R.; Sheu, M.; Kim, N.; Kim, S.; Islam, N.; Wier, E.M.; Wang, G.; Li, A.; Park, A.; et al. Noncoding dsRNA induces retinoic acid synthesis to stimulate hair follicle regeneration via TLR3. Nat. Commun. 2019, 10, 2811. [Google Scholar] [CrossRef] [Green Version]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, A.; Conrad, C. Psoriasis: Classical vs. Paradoxical. The Yin-Yang of TNF and Type I Interferon. Front. Immunol. 2018, 9, 2746. [Google Scholar] [CrossRef]
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kanbe, A.; Ito, H.; Seishima, M. Activation of STING signaling accelerates skin wound healing. J. Derm. Sci. 2020, 97, 21–29. [Google Scholar] [CrossRef] [PubMed]
- de Koning, H.D.; Bergboer, J.G.; van den Bogaard, E.H.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; Simon, A.; Zeeuwen, P.L.; Schalkwijk, J. Strong induction of AIM2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp. Derm. 2012, 21, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Matsuzaki, Y.; Nishikawa, Y.; Kitamura, H.; Akasaka, E.; Rokunohe, D.; Nakano, H.; Imaizumi, T.; Satoh, K.; Sawamura, D. Characterization of retinoic acid-inducible gene-I (RIG-I) expression corresponding to viral infection and UVB in human keratinocytes. J. Derm. Sci. 2012, 66, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [Green Version]
- Tüzün, Y.; Antonov, M.; Dolar, N.; Wolf, R. Keratinocyte cytokine and chemokine receptors. Derm. Clin. 2007, 25, 467–476. [Google Scholar] [CrossRef]
- Krutmann, J.; Czech, W.; Parlow, F.; Trefzer, U.; Kapp, A.; Schöpf, E.; Luger, T.A. Ultraviolet radiation effects on human keratinocyte ICAM-1 expression: UV-induced inhibition of cytokine-induced ICAM-1 mRNA expression is transient, differentially restored for IFN gamma versus TNF alpha, and followed by ICAM-1 induction via a TNF alpha-like pathway. J. Investig. Derm. 1992, 98, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Cavani, A.; Girolomoni, G. Interferon-gamma-stimulated human keratinocytes express the genes necessary for the production of peptide-loaded MHC class II molecules. J. Investig. Derm. 1998, 110, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Hänel, K.H.; Cornelissen, C.; Lüscher, B.; Baron, J.M. Cytokines and the skin barrier. Int. J. Mol. Sci. 2013, 14, 6720–6745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound Care 2016, 5, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-P.; Schunck, M.; Kallen, K.-J.; Neumann, C.; Trautwein, C.; Rose-John, S.; Proksch, E. The interleukin-6 cytokine system regulates epidermal permeability barrier homeostasis. J. Investig. Derm. 2004, 123, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019, 4, 9657. [Google Scholar] [CrossRef] [PubMed]
- Kroeze, K.L.; Boink, M.A.; Sampat-Sardjoepersad, S.C.; Waaijman, T.; Scheper, R.J.; Gibbs, S. Autocrine regulation of re-epithelialization after wounding by chemokine receptors CCR1, CCR10, CXCR1, CXCR2, and CXCR3. J. Investig. Derm. 2012, 132, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv. Wound Care 2013, 2, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Bergers, G. Tumors vs. Chronic Wounds: An Immune Cell’s Perspective. Front. Immunol. 2019, 10, 2178. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R. Growth factors and chronic wound healing: Past, present, and future. Adv. Skin Wound Care 2004, 17, 24–35. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Vallejo, A.F.; Davies, J.; Sirvent, S.; Polak, M.E. Langerhans Cells-Programmed by the Epidermis. Front. Immunol. 2017, 8, 1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botting, R.A.; Haniffa, M. The developing immune network in human prenatal skin. Immunology 2020, 160, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Deckers, J.; Hammad, H.; Hoste, E. Langerhans Cells: Sensing the Environment in Health and Disease. Front. Immunol. 2018, 9, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strbo, N.; Pastar, I.; Romero, L.; Chen, V.; Vujanac, M.; Sawaya, A.P.; Jozic, I.; Ferreira, A.D.F.; Wong, L.L.; Head, C.; et al. Single cell analyses reveal specific distribution of anti-bacterial molecule Perforin-2 in human skin and its modulation by wounding and Staphylococcus aureus infection. Exp. Derm. 2019, 28, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.R.; West, H.C.; Henderson, S.; Ushakov, D.S.; Santos, E.; Sousa, P.; Strid, J.; Chakraverty, R.; Yates, A.J.; Bennett, C.L. A wave of monocytes is recruited to replenish the long-term Langerhans cell network after immune injury. Sci. Immunol. 2019, 4, 8704. [Google Scholar] [CrossRef]
- Seré, K.; Baek, J.-H.; Ober-Blöbaum, J.; Müller-Newen, G.; Tacke, F.; Yokota, Y.; Zenke, M.; Hieronymus, T. Two distinct types of Langerhans cells populate the skin during steady state and inflammation. Immunity 2012, 37, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Gong, S.-J.; Xu, Y.-H.; Hambly, B.D.; Bao, S. Impaired cutaneous wound healing in granulocyte/macrophage colony-stimulating factor knockout mice. Br. J. Derm. 2007, 157, 458–465. [Google Scholar] [CrossRef]
- Mann, A.; Breuhahn, K.; Schirmacher, P.; Blessing, M. Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: Stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J. Investig. Derm. 2001, 117, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Sun, H.; Han, C.; Wang, X.; Yu, W. Topically applied rhGM-CSF for the wound healing: A systematic review. Burns 2011, 37, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.W.; Kupper, T.S. T cells and the skin: From protective immunity to inflammatory skin disorders. Nat. Rev. Immunol. 2019, 19, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kobayashi, T.; Sugihara, E.; Yamada, T.; Ikuta, K.; Pittaluga, S.; Saya, H.; Amagai, M.; Nagao, K. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 2015, 21, 1272–1279. [Google Scholar] [CrossRef]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.-Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8⁺ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Rauschenberger, T.; Schmitt, V.; Azeem, M.; Klein-Hessling, S.; Murti, K.; Grän, F.; Goebeler, M.; Kerstan, A.; Klein, M.; Bopp, T.; et al. T Cells Control Chemokine Secretion by Keratinocytes. Front. Immunol. 2019, 10, 1917. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Bouladoux, N.; Linehan, J.L.; Han, S.-J.; Harrison, O.J.; Wilhelm, C.; Conlan, S.; Himmelfarb, S.; Byrd, A.L.; Deming, C.; et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015, 520, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Linehan, J.L.; Harrison, O.J.; Han, S.-J.; Byrd, A.L.; Vujkovic-Cvijin, I.; Villarino, A.V.; Sen, S.K.; Shaik, J.; Smelkinson, M.; Tamoutounour, S.; et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 2018, 172, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.M.; Witherden, D.A.; Havran, W.L. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol. 2017, 17, 733–745. [Google Scholar] [CrossRef]
- Jung, H.; Hsiung, B.; Pestal, K.; Procyk, E.; Raulet, D.H. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J. Exp. Med. 2012, 209, 2409–2422. [Google Scholar] [CrossRef] [Green Version]
- Whang, M.I.; Guerra, N.; Raulet, D.H. Costimulation of dendritic epidermal gammadelta T cells by a new NKG2D ligand expressed specifically in the skin. J. Immunol. 2009, 182, 4557–4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strid, J.; Roberts, S.J.; Filler, R.B.; Lewis, J.M.; Kwong, B.Y.; Schpero, W.; Kaplan, D.H.; Hayday, A.C.; Girardi, M. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat. Immunol. 2008, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Mohamed, R.H.; Kajikawa, M.; Koizumi, J.; Tanaka, M.; Fugo, K.; Otsuka, N.; Maenaka, K.; Yagita, H.; Chiba, H.; et al. Involvement of an NKG2D ligand H60c in epidermal dendritic T cell-mediated wound repair. J. Immunol. 2012, 188, 3972–3979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jameson, J.M.; Cauvi, G.; Sharp, L.L.; Witherden, D.A.; Havran, W.L. Gammadelta T cell-induced hyaluronan production by epithelial cells regulates inflammation. J. Exp. Med. 2005, 201, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, A.S.; Hemmers, S.; Garijo, O.; Chabod, M.; Mowen, K.; Witherden, D.A.; Havran, W.L. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Investig. 2013, 123, 4364–4374. [Google Scholar] [CrossRef]
- Jameson, J.; Ugarte, K.; Chen, N.; Yachi, P.; Fuchs, E.; Boismenu, R.; Havran, W.L. A role for skin gammadelta T cells in wound repair. Science 2002, 296, 747–749. [Google Scholar] [CrossRef]
- Toulon, A.; Breton, L.; Taylor, K.R.; Tenenhaus, M.; Bhavsar, D.; Lanigan, C.; Rudolph, R.; Jameson, J.; Havran, W.L. A role for human skin-resident T cells in wound healing. J. Exp. Med. 2009, 206, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Than, U.T.T.; Guanzon, D.; Leavesley, D.; Parker, T. Association of Extracellular Membrane Vesicles with Cutaneous Wound Healing. Int. J. Mol. Sci. 2017, 18, 956. [Google Scholar] [CrossRef] [Green Version]
- Than, U.T.T.; Leavesley, D.I.; Parker, T.J. Characteristics and roles of extracellular vesicles released by epidermal keratinocytes. J. Eur. Acad. Derm. Venereol. 2019, 33, 2264–2272. [Google Scholar] [CrossRef]
- Kotzerke, K.; Mempel, M.; Aung, T.; Wulf, G.G.; Urlaub, H.; Wenzel, D.; Schön, M.P.; Braun, A. Immunostimulatory activity of murine keratinocyte-derived exosomes. Exp. Derm. 2013, 22, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Fang, H.; Shao, S.; Dang, E.; Zhang, J.; Qiao, P.; Yang, A.; Wang, G. Keratinocyte exosomes activate neutrophils and enhance skin inflammation in psoriasis. FASEB J. 2019, 33, 13241–13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Brown, B.A.; Siegel, A.P.; El Masry, M.; Zeng, X.; Song, W.; Das, A.; Khandelwal, P.; Clark, A.; Singh, K.; et al. Exosome-Mediated Crosstalk between Keratinocytes and Macrophages in Cutaneous Wound Healing. ACS Nano 2020, 14, 12732–12748. [Google Scholar] [CrossRef] [PubMed]
- Than, U.T.T.; Guanzon, D.; Broadbent, J.A.; Leavesley, D.I.; Salomon, C.; Parker, T.J. Differential Expression of Keratinocyte-Derived Extracellular Vesicle Mirnas Discriminate Exosomes From Apoptotic Bodies and Microvesicles. Front. Endocrinol. 2018, 9, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.R.; Wang, C.; Patel, R.; Trujillo, A.; Patel, N.A.; Prather, J.; Gould, L.J.; Wu, M.H. Human Adipose-Derived Stem Cell Conditioned Media and Exosomes Containing MALAT1 Promote Human Dermal Fibroblast Migration and Ischemic Wound Healing. Adv. Wound Care 2018, 7, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Luan, S.; Chen, J.; Zhou, Y.; Wang, T.; Li, Z.; Fu, Y.; Zhai, A.; Bi, C. The MSC-Derived Exosomal lncRNA H19 Promotes Wound Healing in Diabetic Foot Ulcers by Upregulating PTEN via MicroRNA-152-3p. Mol. Ther. Nucleic Acids 2020, 19, 814–826. [Google Scholar] [CrossRef]
- Shabbir, A.; Cox, A.; Rodriguez-Menocal, L.; Salgado, M.; Van Badiavas, E. Mesenchymal Stem Cell Exosomes Induce Proliferation and Migration of Normal and Chronic Wound Fibroblasts, and Enhance Angiogenesis In Vitro. Stem Cells Dev. 2015, 24, 1635–1647. [Google Scholar] [CrossRef]
- Tao, S.-C.; Guo, S.-C.; Li, M.; Ke, Q.-F.; Guo, Y.-P.; Zhang, C.-Q. Chitosan Wound Dressings Incorporating Exosomes Derived from MicroRNA-126-Overexpressing Synovium Mesenchymal Stem Cells Provide Sustained Release of Exosomes and Heal Full-Thickness Skin Defects in a Diabetic Rat Model. Stem Cells Transl. Med. 2017, 6, 736–747. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bai, X.; Zhao, B.; Li, Y.; Zhang, Y.; Li, Z.; Wang, X.; Luo, L.; Han, F.; Zhang, J.; et al. Cell-free therapy based on adipose tissue stem cell-derived exosomes promotes wound healing via the PI3K/Akt signaling pathway. Exp. Cell Res. 2018, 370, 333–342. [Google Scholar] [CrossRef]
- Geiger, A.; Walker, A.; Nissen, E. Human fibrocyte-derived exosomes accelerate wound healing in genetically diabetic mice. Biochem. Biophys. Res. Commun. 2015, 467, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rao, S.S.; Wang, Z.X.; Cao, J.; Tan, Y.J.; Luo, J.; Li, H.M.; Zhang, W.S.; Chen, C.Y.; Xie, H. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through miR-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 2018, 8, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Shiekh, P.A.; Singh, A.; Kumar, A. Exosome laden oxygen releasing antioxidant and antibacterial cryogel wound dressing OxOBand alleviate diabetic and infectious wound healing. Biomaterials 2020, 249, 120020. [Google Scholar] [CrossRef] [PubMed]
- Ti, D.; Hao, H.; Tong, C.; Liu, J.; Dong, L.; Zheng, J.; Zhao, Y.; Liu, H.; Fu, X.; Han, W. LPS-preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome-shuttled let-7b. J. Transl. Med. 2015, 13, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubock, J.; Romani, N.; Grubauer, G.; Fritsch, P. HLA-DR expression on keratinocytes is a common feature of diseased skin. Br. J. Derm. 1986, 114, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Lifshitz, B.; Fu, S.M.; Staiano-Coico, L.; Wang, C.Y.; Carter, D.M. Expression of HLA-DR molecules by keratinocytes, and presence of Langerhans cells in the dermal infiltrate of active psoriatic plaques. J. Exp. Med. 1986, 164, 1013–1028. [Google Scholar] [CrossRef]
- Wikner, N.E.; Huff, J.C.; Norris, D.A.; Boyce, S.T.; Cary, M.; Kissinger, M.; Weston, W.L. Study of HLA-DR synthesis in cultured human keratinocytes. J. Investig. Derm. 1986, 87, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Bal, V.; McIndoe, A.; Denton, G.; Hudson, D.; Lombardi, G.; Lamb, J.; Lechler, R. Antigen presentation by keratinocytes induces tolerance in human T cells. Eur. J. Immunol. 1990, 20, 1893–1897. [Google Scholar] [CrossRef]
- Gaspari, A.A.; Katz, S.I. Induction of in vivo hyporesponsiveness to contact allergens by hapten-modified Ia+ keratinocytes. J. Immunol. 1991, 147, 4155–4161. [Google Scholar]
- Goodman, R.E.; Nestle, F.; Naidu, Y.M.; Green, J.M.; Thompson, C.B.; Nickoloff, B.J.; Turka, L.A. Keratinocyte-derived T cell costimulation induces preferential production of IL-2 and IL-4 but not IFN-gamma. J. Immunol. 1994, 152, 5189–5198. [Google Scholar]
- Carr, M.M.; McVittie, E.; Guy, K.; Gawkrodger, D.J.; Hunter, J.A. MHC class II antigen expression in normal human epidermis. Immunology 1986, 59, 223–227. [Google Scholar] [PubMed]
- Tamoutounour, S.; Han, S.J.; Deckers, J.; Constantinides, M.G.; Hurabielle, C.; Harrison, O.J.; Bouladoux, N.; Linehan, J.L.; Link, V.M.; Vujkovic-Cvijin, I.; et al. Keratinocyte-intrinsic MHCII expression controls microbiota-induced Th1 cell responses. Proc. Natl. Acad. Sci. USA 2019, 116, 23643–23652. [Google Scholar] [CrossRef] [PubMed]
- Wosen, J.E.; Mukhopadhyay, D.; Macaubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 2144. [Google Scholar] [CrossRef] [PubMed]
- Biton, M.; Haber, A.L.; Rogel, N.; Burgin, G.; Beyaz, S.; Schnell, A.; Ashenberg, O.; Su, C.W.; Smillie, C.; Shekhar, K.; et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 2018, 175, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Canesso, M.C.C.; Vieira, A.T.; Castro, T.B.R.; Schirmer, B.G.A.; Cisalpino, D.; Martins, F.S.; Rachid, M.A.; Nicoli, J.R.; Teixeira, M.M.; Barcelos, L.S. Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J. Immunol. 2014, 193, 5171–5180. [Google Scholar] [CrossRef] [Green Version]
- Di Domizio, J.; Belkhodja, C.; Chenuet, P.; Fries, A.; Murray, T.; Mondéjar, P.M.; Demaria, O.; Conrad, C.; Homey, B.; Werner, S.; et al. The commensal skin microbiota triggers type I IFN-dependent innate repair responses in injured skin. Nat. Immunol. 2020, 21, 1034–1045. [Google Scholar] [CrossRef]
- Li, D.; Wang, W.; Wu, Y.; Ma, X.; Zhou, W.; Lai, Y. Lipopeptide 78 from Staphylococcus epidermidis Activates beta-Catenin to Inhibit Skin Inflammation. J. Immunol. 2019, 202, 1219–1228. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Lei, H.; Li, Z.; Li, H.; Wang, Y.; Lai, Y. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS ONE 2013, 8, e58288. [Google Scholar] [CrossRef] [Green Version]
- Chessa, C.; Bodet, C.; Jousselin, C.; Wehbe, M.; Lévêque, N.; Garcia, M. Antiviral and Immunomodulatory Properties of Antimicrobial Peptides Produced by Human Keratinocytes. Front. Microbiol. 2020, 11, 1155. [Google Scholar] [CrossRef]
- Gläser, R.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schröder, J.-M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2005, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kisich, K.O.; Howell, M.D.; Boguniewicz, M.; Heizer, H.R.; Watson, N.U.; Leung, D.Y.M. The constitutive capacity of human keratinocytes to kill Staphylococcus aureus is dependent on beta-defensin 3. J. Investig. Derm. 2007, 127, 2368–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Cogen, A.L.; Radek, K.A.; Park, H.J.; Macleod, D.T.; Leichtle, A.; Ryan, A.F.; Di Nardo, A.; Gallo, R.L. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J. Investig. Derm. 2010, 130, 2211–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, 4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyerich, S.; Eyerich, K.; Traidl-Hoffmann, C.; Biedermann, T. Cutaneous Barriers and Skin Immunity: Differentiating a Connected Network. Trends Immunol. 2018, 39, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L.; McDermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Derm. 2016, 25, 167–173. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Heilborn, J.D.; Nilsson, M.F.; Kratz, G.; Weber, G.; Sørensen, O.; Borregaard, N.; Ståhle-Bäckdahl, M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J. Investig. Derm. 2003, 120, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Santiago, B.; Trujillo, V.; Montoya, A.; Gonzalez-Curiel, I.; Castañeda-Delgado, J.; Cardenas, A.; Rincon, K.; Hernandez, M.L.; Hernández-Pando, R. Expression of antimicrobial peptides in diabetic foot ulcer. J. Derm. Sci. 2012, 65, 19–26. [Google Scholar] [CrossRef]
- Carretero, M.; Escámez, M.J.; García, M.; Duarte, B.; Holguín, A.; Retamosa, L.; Jorcano, J.L.; Río, M.D.; Larcher, F. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J. Investig. Derm. 2008, 128, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.J.; Stevenson, A.; Fear, M.W.; Wood, F.M. A review of epigenetic regulation in wound healing: Implications for the future of wound care. Wound Repair Regen. 2020, 28, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meyerson, M. Illuminating the noncoding genome in cancer. Nat. Cancer 2020, 1, 864–872. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hodgkinson, T.; Gothard, E.J.; Boroumand, S.; Lamb, R.; Cummins, I.; Narang, P.; Sawtell, A.; Coles, J.; Leonov, G.; et al. Epidermal Notch1 recruits RORγ(+) group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun. 2016, 7, 11394. [Google Scholar] [CrossRef]
- Na, J.; Shin, J.Y.; Jeong, H.; Lee, J.Y.; Kim, B.J.; Kim, W.S.; Yune, T.Y.; Ju, B.-G. JMJD3 and NF-κB-dependent activation of Notch1 gene is required for keratinocyte migration during skin wound healing. Sci. Rep. 2017, 7, 6494. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Hamada, A.; Torre, C.; Drancourt, M.; Ghigo, E. Trained Immunity Carried by Non-immune Cells. Front. Microbiol. 2018, 9, 3225. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Larsen, S.B.; Gomez, N.C.; Alaverdyan, K.; Sendoel, A.; Yuan, S.; Polak, L.; Kulukian, A.; Chai, S.; Fuchs, E. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 2017, 550, 475–480. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Lee, R.C.; Lavanway, A.; Williams, P.T.; Jewell, D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr. Biol. 2003, 13, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herter, E.K.; Xu Landén, N. Non-Coding RNAs: New Players in Skin Wound Healing. Adv. Wound Care 2017, 6, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, E.J.; Dunne, N.; McCarthy, H.O. MicroRNA as Therapeutic Targets for Chronic Wound Healing. Mol. Ther. Nucleic Acids 2017, 8, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, A.M.; Das, S.; Abd Ghafar, N.; Teoh, S.L. Role of MicroRNA in Proliferation Phase of Wound Healing. Front. Genet. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Lang, H.; Wang, Z.; Zhang, T.; Zhang, D.; Wang, R.; Lin, X.; Liu, X.; Shi, P.; Pang, X. Human Novel MicroRNA Seq-915_x4024 in Keratinocytes Contributes to Skin Regeneration by Suppressing Scar Formation. Mol. Ther. Nucleic Acids 2019, 14, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.; Zhao, F.; Zhang, T.; Liu, X.; Wang, Z.; Wang, R.; Shi, P.; Pang, X. MicroRNA-149 contributes to scarless wound healing by attenuating inflammatory response. Mol. Med. Rep. 2017, 16, 2156–2162. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, Y. Resveratrol alleviates LPS-induced injury in human keratinocyte cell line HaCaT by up-regulation of miR-17. Biochem. Biophys. Res. Commun. 2018, 501, 106–112. [Google Scholar] [CrossRef]
- Li, H.; Han, X.; Zuo, K.; Li, L.; Liu, J.; Yuan, X.; Shen, Y.; Shao, M.; Pang, D.; Chu, Y.; et al. miR-23b promotes cutaneous wound healing through inhibition of the inflammatory responses by targeting ASK1. Acta Biochim. Biophys. Sin. 2018, 50, 1104–1113. [Google Scholar] [CrossRef]
- Li, D.; Li, X.I.; Wang, A.; Meisgen, F.; Pivarcsi, A.; Sonkoly, E.; Stahle, M.; Landen, N.X. MicroRNA-31 Promotes Skin Wound Healing by Enhancing Keratinocyte Proliferation and Migration. J. Investig. Derm. 2015, 135, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Ma, X.; Su, Y.; Song, Y.; Tian, Y.; Yuan, S.; Zhang, X.; Yang, D.; Zhang, H.; Shuai, J.; et al. MiR-31 Mediates Inflammatory Signaling to Promote Re-Epithelialization during Skin Wound Healing. J. Investig. Derm. 2018, 138, 2253–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Pal, A.S.; Hsu, A.Y.-H.; Gurol, T.; Zhu, X.; Wirbisky-Hershberger, S.E.; Freeman, J.L.; Kasinski, A.L.; Deng, Q. MicroRNA-223 Suppresses the Canonical NF-κB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell. Rep. 2018, 22, 1810–1823. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Peng, H.; Qu, L.; Sommar, P.; Wang, A.; Chu, T.; Li, X.; Bi, X.; Liu, Q.; Serezal, I.G.; et al. miR-19a/b and miR-20a promote wound healing by regulating the inflammatory response of keratinocytes. J. Investig. Derm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, W.; Zhang, L.; Dorset-Martin, W.; Morris, M.W.; Mitchell, M.E.; Liechty, K.W. The role of microRNA-146a in the pathogenesis of the diabetic wound-healing impairment: Correction with mesenchymal stem cell treatment. Diabetes 2012, 61, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, X.; Li, D.; Ren, X.; Li, Y.; Herter, E.K.; Qian, M.; Toma, M.A.; Wintler, A.M.; Serezal, I.G.; et al. MicroRNA-34 Family Enhances Wound Inflammation by Targeting LGR4. J. Investig. Derm. 2020, 140, 465–476. [Google Scholar] [CrossRef]
- Viticchiè, G.; Lena, A.M.; Cianfarani, F.; Odorisio, T.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. MicroRNA-203 contributes to skin re-epithelialization. Cell Death Dis. 2012, 3, e435. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Sun, Y.; Xu, M.; Zeng, F.; Xiong, X. miR-203 Acts as an Inhibitor for Epithelial-Mesenchymal Transition Process in Diabetic Foot Ulcers via Targeting Interleukin-8. Neuroimmunomodulation 2019, 26, 239–249. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J.; Guo, S.L.; Fan, K.J.; Li, J.; Wang, Y.L.; Teng, Y.; Yang, X. miR-21 promotes keratinocyte migration and re-epithelialization during wound healing. Int. J. Biol. Sci. 2011, 7, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Pastar, I.; Khan, A.A.; Stojadinovic, O.; Lebrun, E.A.; Medina, M.C.; Brem, H.; Kirsner, R.S.; Jimenez, J.J.; Leslie, C.; Tomic-Canic, M. Induction of specific microRNAs inhibits cutaneous wound healing. J. Biol. Chem. 2012, 287, 29324–29335. [Google Scholar] [CrossRef] [Green Version]
- Hadjicharalambous, M.R.; Lindsay, M.A. Long Non-Coding RNAs and the Innate Immune Response. Non-Coding RNA 2019, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonkoly, E.; Bata-Csorgo, Z.; Pivarcsi, A.; Polyanka, H.; Kenderessy-Szabo, A.; Molnar, G.; Szentpali, K.; Bari, L.; Megyeri, K.; Mandi, Y.; et al. Identification and characterization of a novel, psoriasis susceptibility-related noncoding RNA gene, PRINS. J. Biol. Chem. 2005, 280, 24159–24167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danis, J.; Göblös, A.; Bata-Csörgő, Z.; Kemény, L.; Széll, M. PRINS Non-Coding RNA Regulates Nucleic Acid-Induced Innate Immune Responses of Human Keratinocytes. Front. Immunol. 2017, 8, 1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herter, E.K.; Li, D.; Toma, M.A.; Vij, M.; Li, X.; Visscher, D.; Wang, A.; Chu, T.; Sommar, P.; Blomqvist, L.; et al. WAKMAR2, a Long Noncoding RNA Downregulated in Human Chronic Wounds, Modulates Keratinocyte Motility and Production of Inflammatory Chemokines. J. Investig. Derm. 2019, 139, 1373–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, A.; Jan, A.; Wajid, M.A.; Tariq, S. Management of Chronic Non-healing Wounds by Hirudotherapy. World J. Plast. Surg. 2017, 6, 9–17. [Google Scholar] [PubMed]
- Jarbrink, K.; Ni, G.; Sonnergren, H.; Schmidtchen, A.; Pang, C.; Bajpai, R.; Car, J. The humanistic and economic burden of chronic wounds: A protocol for a systematic review. Syst. Rev. 2017, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Grone, A. Keratinocytes and cytokines. Vet. Immunol. Immunopathol. 2002, 88, 1–12. [Google Scholar] [CrossRef]
- Lan, C.C.; Wu, C.S.; Huang, S.M.; Wu, I.H.; Chen, G.S. High-glucose environment enhanced oxidative stress and increased interleukin-8 secretion from keratinocytes: New insights into impaired diabetic wound healing. Diabetes 2013, 62, 2530–2538. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.J.; Yager, D.R.; Pozez, A.L.; Olutoye, O.O.; Crossland, M.C.; Diegelmann, R.F.; Cohen, I.K. Interleukin-1alpha and collagenase activity are elevated in chronic wounds. Plast. Reconstr. Surg. 1998, 102, 1023–1027, discussion 1028–1029. [Google Scholar] [CrossRef]
- Cowin, A.J.; Hatzirodos, N.; Rigden, J.; Fitridge, R.; Belford, D.A. Etanercept decreases tumor necrosis factor-alpha activity in chronic wound fluid. Wound Repair Regen. 2006, 14, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Luckett-Chastain, L.R.; Calhoun, K.N.; Frempah, B.; Bastian, A.; Gallucci, R.M. Interleukin 6 Function in the Skin and Isolated Keratinocytes Is Modulated by Hyperglycemia. J. Immunol. Res. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, R.J. Chemokine Regulation of Angiogenesis During Wound Healing. Adv. Wound Care 2015, 4, 641–650. [Google Scholar] [CrossRef]
- Dasu, M.R.; Martin, S.J. Toll-like receptor expression and signaling in human diabetic wounds. World J. Diabetes 2014, 5, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Agrawal, N.K.; Gupta, S.K.; Sinha, P.; Singh, K. Increased expression of TLR9 associated with pro-inflammatory S100A8 and IL-8 in diabetic wounds could lead to unresolved inflammation in type 2 diabetes mellitus (T2DM) cases with impaired wound healing. J. Diabetes Complicat. 2016, 30, 99–108. [Google Scholar] [CrossRef]
- Singh, K.; Singh, V.K.; Agrawal, N.K.; Gupta, S.K.; Singh, K. Association of Toll-like receptor 4 polymorphisms with diabetic foot ulcers and application of artificial neural network in DFU risk assessment in type 2 diabetes patients. Biomed. Res. Int 2013, 2013, 318686. [Google Scholar] [CrossRef]
- Wifi, M.A.; Assem, M.; Elsherif, R.H.; El-Azab, H.A.; Saif, A. Toll-like receptors-2 and -9 (TLR2 and TLR9) gene polymorphism in patients with type 2 diabetes and diabetic foot. Medicine 2017, 96, e6760. [Google Scholar] [CrossRef]
- Dasu, M.R.; Thangappan, R.K.; Bourgette, A.; DiPietro, L.A.; Isseroff, R.; Jialal, I. TLR2 expression and signaling-dependent inflammation impair wound healing in diabetic mice. Lab. Investig. 2010, 90, 1628–1636. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Quan, Y.; Liu, Y.; Liu, K.; Li, H.; Jiang, Z.; Zhang, T.; Lei, H.; Radek, K.A.; Li, D.; et al. Hyperglycaemia inhibits REG3A expression to exacerbate TLR3-mediated skin inflammation in diabetes. Nat. Commun. 2016, 7, 13393. [Google Scholar] [CrossRef]
- Dasu, M.R.; Jialal, I. Amelioration in wound healing in diabetic toll-like receptor-4 knockout mice. J. Diabetes Complicat. 2013, 27, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Dressel, S.; Harder, J.; Cordes, J.; Wittersheim, M.; Meyer-Hoffert, U.; Sunderkotter, C.; Glaser, R. Differential expression of antimicrobial peptides in margins of chronic wounds. Exp. Derm. 2010, 19, 628–632. [Google Scholar] [CrossRef]
- Thorey, I.S.; Roth, J.; Regenbogen, J.; Halle, J.P.; Bittner, M.; Vogl, T.; Kaesler, S.; Bugnon, P.; Reitmaier, B.; Durka, S.; et al. The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J. Biol. Chem. 2001, 276, 35818–35825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronberg, A.; Mahlapuu, M.; Stahle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL-37 is safe and effective in enhancing healing of hard-to-heal venous leg ulcers: A randomized, placebo-controlled clinical trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Drago, F.; Gariazzo, L.; Cioni, M.; Trave, I.; Parodi, A. The microbiome and its relevance in complex wounds. Eur. J. Derm. 2019, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Lee, J.O.; Nakamura, K.; Suzuki, S.; Hendon, D.N.; Kobayashi, M.; Suzuki, F. Lineage -CD34+CD31+ cells that appear in association with severe burn injury are inhibitory on the production of antimicrobial peptides by epidermal keratinocytes. PLoS ONE 2014, 9, e82926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trøstrup, H.; Lerche, C.J.; Christophersen, L.J.; Thomsen, K.; Jensen, P.Ø.; Hougen, H.P.; Høiby, N.; Moser, C. Chronic Pseudomonas aeruginosa biofilm infection impairs murine S100A8/A9 and neutrophil effector cytokines-implications for delayed wound closure? Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Dias, K.; Barbugli, P.A.; de Patto, F.; Lordello, V.B.; de Aquino Penteado, L.; Medeiros, A.I.; Vergani, C.E. Soluble factors from biofilm of Candida albicans and Staphylococcus aureus promote cell death and inflammatory response. BMC Microbiol. 2017, 17, 146. [Google Scholar] [CrossRef]
- Li, X.; Li, D.; Wang, A.; Chu, T.; Lohcharoenkal, W.; Zheng, X.; Grunler, J.; Narayanan, S.; Eliasson, S.; Herter, E.K.; et al. MicroRNA-132 with Therapeutic Potential in Chronic Wounds. J. Investig. Derm. 2017, 137, 2630–2638. [Google Scholar] [CrossRef] [Green Version]
- Meisgen, F.; Xu Landen, N.; Wang, A.; Rethi, B.; Bouez, C.; Zuccolo, M.; Gueniche, A.; Stahle, M.; Sonkoly, E.; Breton, L.; et al. MiR-146a negatively regulates TLR2-induced inflammatory responses in keratinocytes. J. Investig. Derm. 2014, 134, 1931–1940. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shu, B.; Zhou, Z.; Xu, Y.; Liu, Y.; Wang, P.; Xiong, K.; Xie, J. Involvement of miRNA203 in the proliferation of epidermal stem cells during the process of DM chronic wound healing through Wnt signal pathways. Stem Cell Res. Ther. 2020, 11, 348. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Keratinocyte Pattern-Recognition Receptors | Pathogen-Associated Molecular Patterns | Keratinocyte-Derived Inflammatory Mediators | References |
|---|---|---|---|
| TLR1/TLR2 | Bacterial tri-acylated lipoproteins | Chemokine (C-C motif) ligand 20 (CCL20), CCL2, interleukin 18 (IL-8) | [14] |
| TLR2/6 | Staphylococcus aureus peptidoglycan and lipoteichoic acid | IL-8, inducible nitric oxide synthase (iNOS) | [15] |
| TLR3 | Double-strand RNA from viruses or damaged cells | Interferon Beta (IFN-β), IL-8, IL-18, Tumor necrosis factor (TNFα), IL-36γ, Chemokine (C-X-C motif) ligand 9 (CXCL9), CCL2, CCL20, CCL27 | [9,16,17,18] |
| TLR4 | Bacterial lipopolysaccharide | IL-1β, TNF-α, IL-8, CCL2, CCL20 | [9] |
| TLR5 | Bacterial flagellin | IL-8, TNF-α, CCL2, CCL20, CCL27 | [9,16] |
| TLR9 | Bacterial unmethylated CpG-containing DNA | TNF-α, IL-8, CXCL10, CCL2, CCL20 | [9,12] |
| MicroRNAs | Functions | Major Targets | References |
|---|---|---|---|
| hsa-miR-132-3p | Reduces keratinocyte-derived chemokines and cytokines while promoting keratinocyte proliferation and migration | HB-EGF 1 | [127] |
| Seq-915_x4024 | Inhibits keratinocyte-derived chemokines and cytokines while enhancing the proliferation of keratinocytes and their ability to promote fibroblast migration and growth | Sar1A, Smad2, TNF-α 2, and IL-8 3 | [137] |
| hsa-miR-149-5p | Downregulates keratinocyte inflammatory cytokine expression | IL-1α, IL-1β, and IL-6 | [138] |
| hsa-miR-17-5p | Downregulates keratinocyte inflammatory cytokine expression | Not shown | [139] |
| hsa-miR-23b-3p | Inhibits keratinocyte-derived pro-inflammatory cytokines | ASK1 4 | [140] |
| hsa-miR-31-5p | Promotes keratinocyte proliferation and migration | EMP-1 5 | [141] |
| mmu-miR-31-5p | Promotes keratinocyte proliferation and migration | Rasa1 6, Spred1 7, Spred2, and Spry4 8 | [142] |
| dre-mir-223 | Suppresses NF-κB 9 activation in basal epithelial cells to dampen neutrophil recruitment and inflammation | Cul1a 10, Cul1b, Traf611, and Tab1 12 | [143] |
| hsa-miR-19a-3p and hsa-miR-19b-3p | Decreases TLR3 13-mediated NF-κB activation in keratinocytes | SHCBP1 14 | [144] |
| hsa-miR-20a-5p | Decreases TLR3-mediated NF-κB activation in keratinocytes | SEMA7A 15 | [144] |
| hsa-miR-146a-5p | Inhibits the NF-κB signaling pathway in keratinocytes | IRAK1 16 and TRAF6 | [145] |
| hsa-miR-34a-5p and hsa-miR-34c-5p | Promotes inflammatory chemokine and cytokine production by keratinocytes | LGR4 17 | [146] |
| hsa-miR-203a-3p | Suppresses skin re-epithelialization | RAN 18, RAPH1 19, and IL-8 | [147,148] |
| hsa-miR-21-5p | Promotes skin re-epithelialization | TIMP3 20 and TIAM1 21 | [149] |
| hsa-miR-130a-3p | Suppresses skin re-epithelialization | LepR 22 | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piipponen, M.; Li, D.; Landén, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. https://doi.org/10.3390/ijms21228790
Piipponen M, Li D, Landén NX. The Immune Functions of Keratinocytes in Skin Wound Healing. International Journal of Molecular Sciences. 2020; 21(22):8790. https://doi.org/10.3390/ijms21228790
Chicago/Turabian StylePiipponen, Minna, Dongqing Li, and Ning Xu Landén. 2020. "The Immune Functions of Keratinocytes in Skin Wound Healing" International Journal of Molecular Sciences 21, no. 22: 8790. https://doi.org/10.3390/ijms21228790
APA StylePiipponen, M., Li, D., & Landén, N. X. (2020). The Immune Functions of Keratinocytes in Skin Wound Healing. International Journal of Molecular Sciences, 21(22), 8790. https://doi.org/10.3390/ijms21228790