Interactions and Signal Transduction Pathways Involved during Central Nervous System Entry by Neisseria meningitidis across the Blood–Brain Barriers


Abstract
:1. Introduction
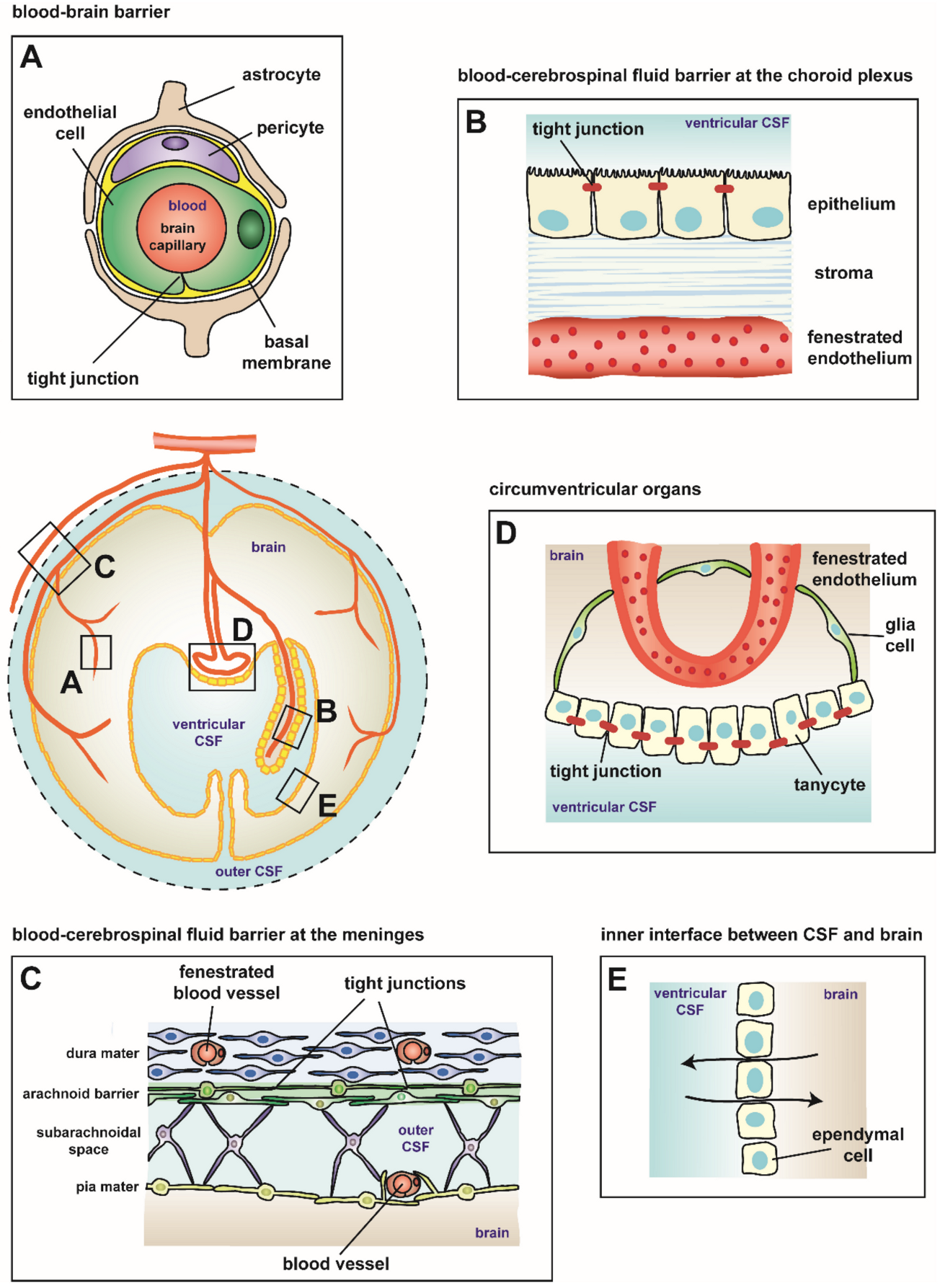
2. Blood–Brain Barriers
2.1. Blood–Brain Barrier
2.2. Blood–Cerebrospinal Fluid Barrier at the Choroid Plexus
2.3. Blood–Cerebrospinal Fluid Barrier at the Meninges
3. Neisserial Interactions with the Host
3.1. Colonization as a Prerequisite for Meningococcal Disease
Minor Adhesins and Invasins Involved during Interaction with Epithelial Cells
3.2. Neisserial Survival in the Blood
3.3. Neisserial Interactions with the Host during CNS Entry
3.3.1. Neisserial Interactions with the Blood–Brain Barrier
3.3.2. Neisserial Interactions with the Blood–Cerebrospinal Fluid Barrier at the Choroid Plexus
3.3.3. Neisserial Interactions with the Blood–Cerebrospinal Fluid Barrier at the Meninges
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AJ | Adherens junction |
| APC | Antigen presenting cell |
| ASM | Acid sphingomyelinase |
| β2AR | β2-adrenergic receptor |
| BBB | Blood–brain barrier |
| BCSFB | Blood–cerebrospinal fluid barrier |
| CEACAM | Carcinoembryonic antigen-related cell adhesion molecules |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CVO | Circumventricular organ |
| ECM | Extracellular matrix |
| FAK | Focal adhesion kinase |
| GDF | Growth differentiation factor |
| GLUT1 | Glucose transporter 1 |
| HSPG | Heparin sulfate proteoglycan |
| IMD | Invasive meningococcal disease |
| JAM | Junction adhesion molecule |
| LPS | Lipopolysaccharide |
| MAGUK | Membrane-associated guanylate kinase |
| MAPK | Mitogen-activated kinase |
| MARVELD3 | MARVEL domain-containing protein 3 |
| MHC | Major histocompatibility complex |
| MLST | Multi locus sequence typing |
| MMP | Matrix metalloprotease |
| MRT | Magnetic resonance tomography |
| N. meningitidis | Neisseria mengitidis |
| ORF | Open reading frame |
| PC-PLC | Phosphatidylcholine specific phospholipase C |
| pdhC | Pyruvate dehydrogenase subunit |
| PECAM | Platelet endothelial cell adhesion molecule |
| PLVAP | Plasmalemma vesicle-associated protein |
| Pgp | P-glycoprotein |
| PRR | Pattern recognition receptor |
| SAS | Subarachnoidal space |
| SLC | Solute carrier |
| ssm | Single strand mispairing |
| TAMP | Tight junction–associated MARVEL protein |
| TEER | Transepithelial electrical resistance |
| TJ | Tight junction |
| Tfp | Type IV pilus |
| TLR | Toll like receptor |
| VE-cadherin | vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| ZO | Zonula occludens |
References
- Rouphael, N.G.; Stephens, D.S. Neisseria meningitidis: Biology, microbiology, and epidemiology. Methods Mol. Biol. 2012, 799, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coureuil, M.; Jamet, A.; Bille, E.; Lecuyer, H.; Bourdoulous, S.; Nassif, X. Molecular interactions between Neisseria meningitidis and its human host. Cell. Microbiol. 2019, 21, e13063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriani, K.S.; Brouwer, M.C.; Baas, F.; Zwinderman, A.H.; van der Ende, A.; van de Beek, D. Genetic variation in the β2-adrenocepter gene is associated with susceptibility to bacterial meningitis in adults. PLoS ONE 2012, 7, e37618. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.C.; de Gans, J.; Heckenberg, S.G.; Zwinderman, A.H.; van der Poll, T.; van de Beek, D. Host genetic susceptibility to pneumococcal and meningococcal disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2009, 9, 31–44. [Google Scholar] [CrossRef]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef] [Green Version]
- Bille, E.; Meyer, J.; Jamet, A.; Euphrasie, D.; Barnier, J.P.; Brissac, T.; Larsen, A.; Pelissier, P.; Nassif, X. A virulence-associated filamentous bacteriophage of Neisseria meningitidis increases host-cell colonisation. PLoS Pathog. 2017, 13, e1006495. [Google Scholar] [CrossRef]
- Bille, E.; Zahar, J.R.; Perrin, A.; Morelle, S.; Kriz, P.; Jolley, K.A.; Maiden, M.C.; Dervin, C.; Nassif, X.; Tinsley, C.R. A chromosomally integrated bacteriophage in invasive meningococci. J. Exp. Med. 2005, 201, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Caugant, D.A.; Tzanakaki, G.; Kriz, P. Lessons from meningococcal carriage studies. FEMS Microbiol. Rev. 2007, 31, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.J.; Griffiths, N.J.; Borodina, E.; Virji, M. Cellular and molecular biology of Neisseria meningitidis colonization and invasive disease. Clin. Sci. (London, England: 1979) 2010, 118, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Peterson, M.E.; Li, Y.; Bita, A.; Moureau, A.; Nair, H.; Kyaw, M.H.; Meningococcal Surveillance, G.; Abad, R.; Bailey, F.; Garcia, I.F.; et al. Meningococcal serogroups and surveillance: A systematic review and survey. J. Glob. Health 2019, 9, 010409. [Google Scholar] [CrossRef]
- Brady, R.C. Meningococcal Infections in Children and Adolescents: Update and Prevention. Adv. Pediatr. 2020, 67, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Møllgård, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghersi-Egea, J.F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, N.R.; Dziegielewska, K.M.; Mollgard, K.; Habgood, M.D. Physiology and molecular biology of barrier mechanisms in the fetal and neonatal brain. J. Physiol. 2018, 596, 5723–5756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef] [PubMed]
- Castro Dias, M.; Mapunda, J.A.; Vladymyrov, M.; Engelhardt, B. Structure and Junctional Complexes of Endothelial, Epithelial and Glial Brain Barriers. Int. J. Mol. Sci. 2019, 20, 5372. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Join-Lambert, O.; Morand, P.C.; Carbonnelle, E.; Coureuil, M.; Bille, E.; Bourdoulous, S.; Nassif, X. Mechanisms of meningeal invasion by a bacterial extracellular pathogen, the example of Neisseria meningitidis. Prog. Neurobiol. 2010, 130–139. [Google Scholar] [CrossRef]
- Christodoulides, M.; Makepeace, B.L.; Partridge, K.A.; Kaur, D.; Fowler, M.I.; Weller, R.O.; Heckels, J.E. Interaction of Neisseria meningitidis with human meningeal cells induces the secretion of a distinct group of chemotactic, proinflammatory, and growth-factor cytokines. Infect. Immun. 2002, 70, 4035–4044. [Google Scholar] [CrossRef] [Green Version]
- Rua, R.; McGavern, D.B. Advances in Meningeal Immunity. Trends Mol. Med. 2018, 24, 542–559. [Google Scholar] [CrossRef]
- Chamot-Rooke, J.; Mikaty, G.; Malosse, C.; Soyer, M.; Dumont, A.; Gault, J.; Imhaus, A.F.; Martin, P.; Trellet, M.; Clary, G.; et al. Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science 2011, 331, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol, C.; Eugene, E.; de Saint Martin, L.; Nassif, X. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect. Immun. 1997, 65, 4836–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassif, X.; Beretti, J.L.; Lowy, J.; Stenberg, P.; O’Gaora, P.; Pfeifer, J.; Normark, S.; So, M. Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3769–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, R.M.; Sim, R.; Goodwin, L.; Winterbotham, M.; Schneider, M.C.; Read, R.C.; Tang, C.M. Identification of meningococcal genes necessary for colonization of human upper airway tissue. Infect. Immun. 2009, 77, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, D.S.; McGee, Z.A. Attachment of Neisseria meningitidis to human mucosal surfaces: Influence of pili and type of receptor cell. J. Infect. Dis. 1981, 143, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.J.; Rifenbery, D.B.; Arvidson, C.G.; So, M. Traversal of a polarized epithelium by pathogenic Neisseriae: Facilitation by type IV pili and maintenance of epithelial barrier function. Mol. Med. 1996, 2, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Kallstrom, H.; Liszewski, M.K.; Atkinson, J.P.; Jonsson, A.B. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol. Microbiol. 1997, 25, 639–647. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.; Rytkonen, A.; Bergman, P.; Albiger, B.; Kallstrom, H.; Hokfelt, T.; Agerberth, B.; Cattaneo, R.; Jonsson, A.B. CD46 in meningococcal disease. Science 2003, 301, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Teuchert, M.; Maisner, A.; Herrler, G. Importance of the carboxyl-terminal FTSL motif of membrane cofactor protein for basolateral sorting and endocytosis. Positive and negative modulation by signals inside and outside the cytoplasmic tail. J. Biol. Chem. 1999, 274, 19979–19984. [Google Scholar] [CrossRef] [Green Version]
- Maisner, A.; Liszewski, M.K.; Atkinson, J.P.; Schwartz-Albiez, R.; Herrler, G. Two different cytoplasmic tails direct isoforms of the membrane cofactor protein (CD46) to the basolateral surface of Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 18853–18858. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.B.; Atkinson, J.P. CD46 in Neisseria pathogenesis. Trends Mol. Med. 2004, 10, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, M.; Heuer, D.; Meyer, T.F. CD46-independent binding of neisserial type IV pili and the major pilus adhesin, PilC, to human epithelial cells. Infect. Immun. 2005, 73, 3072–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchner, M.; Meyer, T.F. The PilC adhesin of the Neisseria type IV pilus-binding specificities and new insights into the nature of the host cell receptor. Mol. Microbiol. 2005, 56, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.C.; Quattroni, P.; Exley, R.M.; Tang, C.M. Transcellular passage of Neisseria meningitidis across a polarized respiratory epithelium. Infect. Immun. 2010, 78, 3832–3847. [Google Scholar] [CrossRef] [Green Version]
- Merz, A.J.; Enns, C.A.; So, M. Type IV pili of pathogenic Neisseriae elicit cortical plaque formation in epithelial cells. Mol. Microbiol. 1999, 32, 1316–1332. [Google Scholar] [CrossRef]
- Merz, A.J.; So, M. Attachment of piliated, Opa- and Opc- gonococci and meningococci to epithelial cells elicits cortical actin rearrangements and clustering of tyrosine-phosphorylated proteins. Infect. Immun. 1997, 65, 4341–4349. [Google Scholar] [CrossRef] [Green Version]
- Stephens, D.S.; Hoffman, L.H.; McGee, Z.A. Interaction of Neisseria meningitidis with human nasopharyngeal mucosa: Attachment and entry into columnar epithelial cells. J. Infect. Dis. 1983, 148, 369–376. [Google Scholar] [CrossRef]
- Deghmane, A.E.; Giorgini, D.; Larribe, M.; Alonso, J.M.; Taha, M.K. Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol. Microbiol. 2002, 43, 1555–1564. [Google Scholar] [CrossRef]
- Spinosa, M.R.; Progida, C.; Talà, A.; Cogli, L.; Alifano, P.; Bucci, C. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect. Immun. 2007, 75, 3594–3603. [Google Scholar] [CrossRef] [Green Version]
- Sjölinder, H.; Jonsson, A.B. Olfactory nerve--a novel invasion route of Neisseria meningitidis to reach the meninges. PLoS ONE 2010, 5, e14034. [Google Scholar] [CrossRef]
- Khairalla, A.S.; Omer, S.A.; Mahdavi, J.; Aslam, A.; Dufailu, O.A.; Self, T.; Jonsson, A.B.; Geörg, M.; Sjölinder, H.; Royer, P.J.; et al. Nuclear trafficking, histone cleavage and induction of apoptosis by the meningococcal App and MspA autotransporters. Cell. Microbiol. 2015, 17, 1008–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadarangani, M.; Pollard, A.J.; Gray-Owen, S.D. Opa proteins and CEACAMs: Pathways of immune engagement for pathogenic Neisseria. FEMS Microbiol. Rev. 2011, 35, 498–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johswich, K.O.; McCaw, S.E.; Islam, E.; Sintsova, A.; Gu, A.; Shively, J.E.; Gray-Owen, S.D. In vivo adaptation and persistence of Neisseria meningitidis within the nasopharyngeal mucosa. PLoS Pathog. 2013, 9, e1003509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slevogt, H.; Zabel, S.; Opitz, B.; Hocke, A.; Eitel, J.; N’Guessan, P.D.; Lucka, L.; Riesbeck, K.; Zimmermann, W.; Zweigner, J.; et al. CEACAM1 inhibits Toll-like receptor 2-triggered antibacterial responses of human pulmonary epithelial cells. Nat. Immunol. 2008, 9, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- De Vries, F.P.; Cole, R.; Dankert, J.; Frosch, M.; van Putten, J.P. Neisseria meningitidis producing the Opc adhesin binds epithelial cell proteoglycan receptors. Mol. Microbiol. 1998, 27, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Virji, M.; Makepeace, K.; Ferguson, D.J.; Achtman, M.; Moxon, E.R. Meningococcal Opa and Opc proteins: Their role in colonization and invasion of human epithelial and endothelial cells. Mol. Microbiol. 1993, 10, 499–510. [Google Scholar] [CrossRef]
- Virji, M.; Makepeace, K.; Ferguson, D.J.; Achtman, M.; Sarkari, J.; Moxon, E.R. Expression of the Opc protein correlates with invasion of epithelial and endothelial cells by Neisseria meningitidis. Mol. Microbiol. 1992, 6, 2785–2795. [Google Scholar] [CrossRef]
- Müller, M.G.; Moe, N.E.; Richards, P.Q.; Moe, G.R. Resistance of Neisseria meningitidis to human serum depends on T and B cell stimulating protein B. Infect. Immun. 2015, 83, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Gómez, J.A.; Criado, M.T.; Ferreirós, C.M. Cooperation between the components of the meningococcal transferrin receptor, TbpA and TbpB, in the uptake of transferrin iron by the 37-kDa ferric-binding protein (FbpA). Res. Microbiol. 1998, 149, 381–387. [Google Scholar] [CrossRef]
- Pettersson, A.; Klarenbeek, V.; van Deurzen, J.; Poolman, J.T.; Tommassen, J. Molecular characterization of the structural gene for the lactoferrin receptor of the meningococcal strain H44/76. Microb. Pathog. 1994, 17, 395–408. [Google Scholar] [CrossRef]
- Schryvers, A.B.; Morris, L.J. Identification and characterization of the human lactoferrin-binding protein from Neisseria meningitidis. Infect. Immun. 1988, 56, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Lewis, L.A.; Dyer, D.W. Identification of an iron-regulated outer membrane protein of Neisseria meningitidis involved in the utilization of hemoglobin complexed to haptoglobin. J. Bacteriol. 1995, 177, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojiljkovic, I.; Hwa, V.; de Saint Martin, L.; O’Gaora, P.; Nassif, X.; Heffron, F.; So, M. The Neisseria meningitidis haemoglobin receptor: Its role in iron utilization and virulence. Mol. Microbiol. 1995, 15, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Serruto, D.; Spadafina, T.; Ciucchi, L.; Lewis, L.A.; Ram, S.; Tontini, M.; Santini, L.; Biolchi, A.; Seib, K.L.; Giuliani, M.M.; et al. Neisseria meningitidis GNA2132, a heparin-binding protein that induces protective immunity in humans. Proc. Natl. Acad. Sci. USA 2010, 107, 3770–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaut, A.G.; Gilbert, J.V.; Artenstein, M.S.; Capra, J.D. Neisseria gonorrhoeae and neisseria meningitidis: Extracellular enzyme cleaves human immunoglobulin A. Science 1975, 190, 1103–1105. [Google Scholar] [CrossRef]
- Jarva, H.; Ram, S.; Vogel, U.; Blom, A.M.; Meri, S. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J. Immunol. (Baltimore, Md. 1950) 2005, 174, 6299–6307. [Google Scholar] [CrossRef] [Green Version]
- Giuntini, S.; Pajon, R.; Ram, S.; Granoff, D.M. Binding of complement factor H to PorB3 and NspA enhances resistance of Neisseria meningitidis to anti-factor H binding protein bactericidal activity. Infect. Immun. 2015, 83, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.L.; Scarselli, M.; Comanducci, M.; Toneatto, D.; Masignani, V. Neisseria meningitidis factor H-binding protein fHbp: A key virulence factor and vaccine antigen. Expert Rev. Vaccines 2015, 14, 841–859. [Google Scholar] [CrossRef] [Green Version]
- Mackinnon, F.G.; Borrow, R.; Gorringe, A.R.; Fox, A.J.; Jones, D.M.; Robinson, A. Demonstration of lipooligosaccharide immunotype and capsule as virulence factors for Neisseria meningitidis using an infant mouse intranasal infection model. Microb. Pathog. 1993, 15, 359–366. [Google Scholar] [CrossRef]
- Geoffroy, M.C.; Floquet, S.; Métais, A.; Nassif, X.; Pelicic, V. Large-scale analysis of the meningococcus genome by gene disruption: Resistance to complement-mediated lysis. Genome Res. 2003, 13, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Beddek, A.J.; Li, M.S.; Kroll, J.S.; Jordan, T.W.; Martin, D.R. Evidence for capsule switching between carried and disease-causing Neisseria meningitidis strains. Infect. Immun. 2009, 77, 2989–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartley, J.S.; Marfin, A.A.; Edupuganti, S.; Liu, L.J.; Cieslak, P.; Perkins, B.; Wenger, J.D.; Stephens, D.S. Capsule switching of Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 1997, 94, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, R.S.; Law, D.K.; Tyler, S.D.; Stephens, G.S.; Bigham, M.; Zollinger, W.D. Potential capsule switching from serogroup Y to B: The characterization of three such Neisseria meningitidis isolates causing invasive meningococcal disease in Canada. Can. J. Infect. Dis. Med. Microbiol. 2005, 16, 171–174. [Google Scholar] [CrossRef]
- Tettelin, H.; Saunders, N.J.; Heidelberg, J.; Jeffries, A.C.; Nelson, K.E.; Eisen, J.A.; Ketchum, K.A.; Hood, D.W.; Peden, J.F.; Dodson, R.J.; et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 2000, 287, 1809–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhill, J.; Achtman, M.; James, K.D.; Bentley, S.D.; Churcher, C.; Klee, S.R.; Morelli, G.; Basham, D.; Brown, D.; Chillingworth, T.; et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 2000, 404, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Meyer, T.F. The repertoire of silent pilus genes in Neisseria gonorrhoeae: Evidence for gene conversion. Cell 1986, 44, 107–115. [Google Scholar] [CrossRef]
- Segal, E.; Hagblom, P.; Seifert, H.S.; So, M. Antigenic variation of gonococcal pilus involves assembly of separated silent gene segments. Proc. Natl. Acad. Sci. USA 1986, 83, 2177–2181. [Google Scholar] [CrossRef] [Green Version]
- Sarkari, J.; Pandit, N.; Moxon, E.R.; Achtman, M. Variable expression of the Opc outer membrane protein in Neisseria meningitidis is caused by size variation of a promoter containing poly-cytidine. Mol. Microbiol. 1994, 13, 207–217. [Google Scholar] [CrossRef]
- Achtman, M. Epidemic spread and antigenic variability of Neisseria meningitidis. Trends Microbiol. 1995, 3, 186–192. [Google Scholar] [CrossRef]
- Hammerschmidt, S.; Müller, A.; Sillmann, H.; Mühlenhoff, M.; Borrow, R.; Fox, A.; van Putten, J.; Zollinger, W.D.; Gerardy-Schahn, R.; Frosch, M. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): Correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 1996, 20, 1211–1220. [Google Scholar] [CrossRef]
- Jennings, M.P.; Srikhanta, Y.N.; Moxon, E.R.; Kramer, M.; Poolman, J.T.; Kuipers, B.; van der Ley, P. The genetic basis of the phase variation repertoire of lipopolysaccharide immunotypes in Neisseria meningitidis. Microbiology 1999, 145 Pt 11, 3013–3021. [Google Scholar] [CrossRef] [Green Version]
- Stern, A.; Brown, M.; Nickel, P.; Meyer, T.F. Opacity genes in Neisseria gonorrhoeae: Control of phase and antigenic variation. Cell 1986, 47, 61–71. [Google Scholar] [CrossRef]
- Frosch, M.; Vogel, U. Structure and Genetics of the Meningococcal Capsule. In Handbook of Meningococcal Disease; Frosch, M., Maiden, M.C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2006; pp. 145–162. [Google Scholar]
- Kahler, C.M.; Stephens, D.S. Genetic basis for biosynthesis, structure, and function of meningococcal lipooligosaccharide (endotoxin). Crit. Rev. Microbiol. 1998, 24, 281–334. [Google Scholar] [CrossRef]
- Tsai, C.M. Molecular mimicry of host structures by lipooligosaccharides of Neisseria meningitidis: Characterization of sialylated and nonsialylated lacto-N-neotetraose (Galbeta1-4GlcNAcbeta1-3Galbeta1-4Glc) structures in lipooligosaccharides using monoclonal antibodies and specific lectins. Adv. Exp. Med. Biol. 2001, 491, 525–542. [Google Scholar] [CrossRef]
- Lécuyer, H.; Nassif, X.; Coureuil, M. Two strikingly different signaling pathways are induced by meningococcal type IV pili on endothelial and epithelial cells. Infect. Immun. 2012, 80, 175–186. [Google Scholar] [CrossRef] [Green Version]
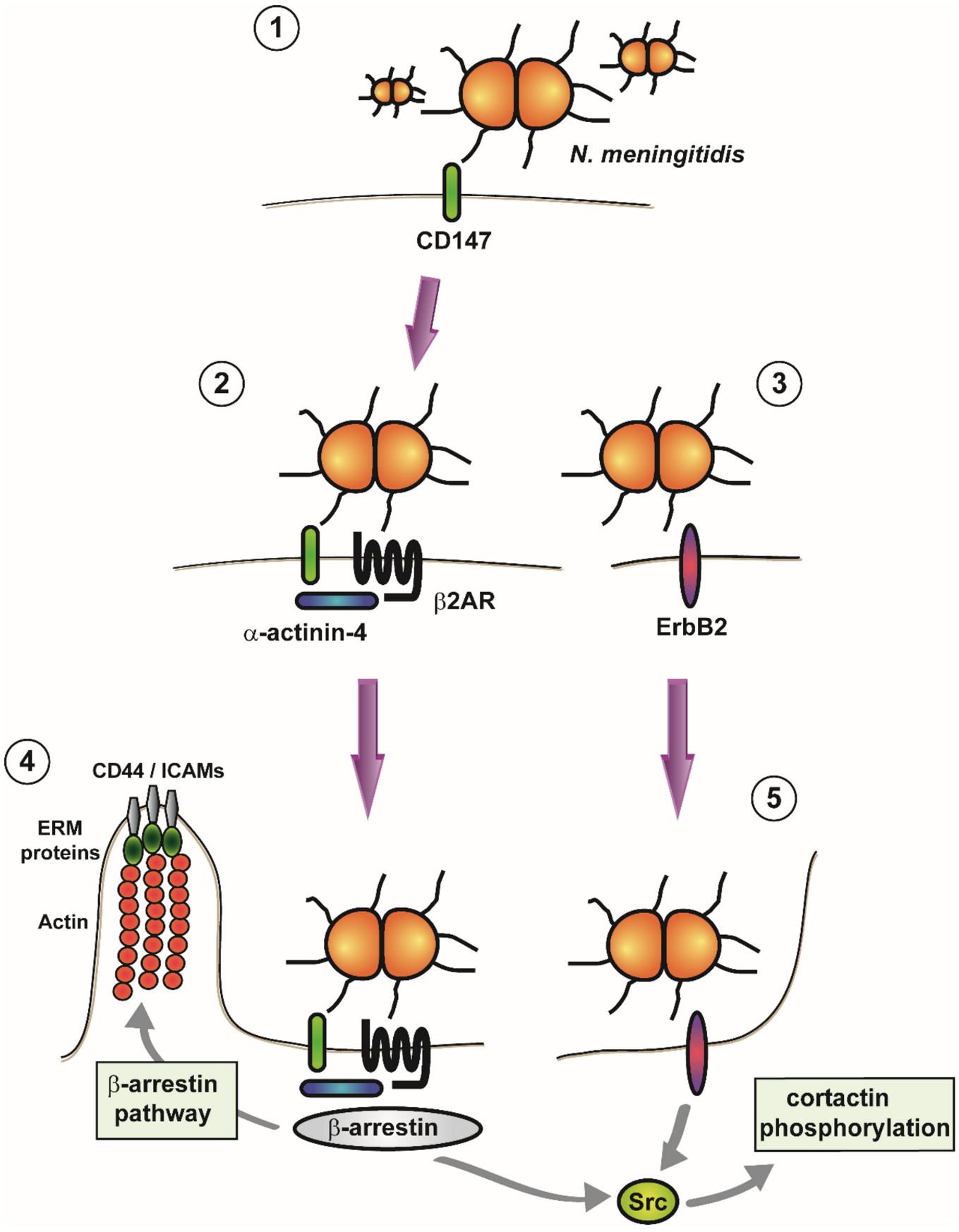
- Bernard, S.C.; Simpson, N.; Join-Lambert, O.; Federici, C.; Laran-Chich, M.P.; Maïssa, N.; Bouzinba-Ségard, H.; Morand, P.C.; Chretien, F.; Taouji, S.; et al. Pathogenic Neisseria meningitidis utilizes CD147 for vascular colonization. Nat. Med. 2014, 20, 725–731. [Google Scholar] [CrossRef]
- Join-Lambert, O.; Lecuyer, H.; Miller, F.; Lelievre, L.; Jamet, A.; Furio, L.; Schmitt, A.; Pelissier, P.; Fraitag, S.; Coureuil, M.; et al. Meningococcal interaction to microvasculature triggers the tissular lesions of purpura fulminans. J. Infect. Dis. 2013, 208, 1590–1597. [Google Scholar] [CrossRef] [Green Version]
- Melican, K.; Michea Veloso, P.; Martin, T.; Bruneval, P.; Duménil, G. Adhesion of Neisseria meningitidis to dermal vessels leads to local vascular damage and purpura in a humanized mouse model. PLoS Pathog. 2013, 9, e1003139. [Google Scholar] [CrossRef] [Green Version]
- Coureuil, M.; Lécuyer, H.; Bourdoulous, S.; Nassif, X. A journey into the brain: Insight into how bacterial pathogens cross blood–brain barriers. Microbiology 2017, 15. [Google Scholar] [CrossRef]
- Kim, B.J.; Schubert-Unkmeir, A. In Vitro Models for Studying the Interaction of Neisseria meningitidis with Human Brain Endothelial Cells. In Neisseria Meningitidis; Humana Press: New York, NY, USA, 2019; Volume 1969, pp. 135–148. [Google Scholar] [CrossRef]
- Melican, K.; Aubey, F.; Duménil, G. Humanized mouse model to study bacterial infections targeting the microvasculature. J. Vis. Exp. JoVE 2014, e51134. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, C.J.; Mahdavi, J.; Thornton, J.; Mann, B.; Wooldridge, K.G.; Abouseada, N.; Oldfield, N.J.; Self, T.; Ala’Aldeen, D.A.; Tuomanen, E.I. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Investig. 2009, 119, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Jen, F.E.; Warren, M.J.; Schulz, B.L.; Power, P.M.; Swords, W.E.; Weiser, J.N.; Apicella, M.A.; Edwards, J.L.; Jennings, M.P. Dual pili post-translational modifications synergize to mediate meningococcal adherence to platelet activating factor receptor on human airway cells. PLoS Pathog. 2013, 9, e1003377. [Google Scholar] [CrossRef] [Green Version]
- Martins Gomes, S.F.; Westermann, A.J.; Sauerwein, T.; Hertlein, T.; Forstner, K.U.; Ohlsen, K.; Metzger, M.; Shusta, E.V.; Kim, B.J.; Appelt-Menzel, A.; et al. Induced Pluripotent Stem Cell-Derived Brain Endothelial Cells as a Cellular Model to Study Neisseria meningitidis Infection. Front. Microbiol. 2019, 10, 1181. [Google Scholar] [CrossRef]
- Maïssa, N.; Covarelli, V.; Janel, S.; Durel, B.; Simpson, N.; Bernard, S.C.; Pardo-Lopez, L.; Bouzinba-Ségard, H.; Faure, C.; Scott, M.G.H.; et al. Strength of Neisseria meningitidis binding to endothelial cells requires highly-ordered CD147/β(2)-adrenoceptor clusters assembled by alpha-actinin-4. Nat. Commun. 2017, 8, 15764. [Google Scholar] [CrossRef]
- Eugene, E.; Hoffmann, I.; Pujol, C.; Couraud, P.O.; Bourdoulous, S.; Nassif, X. Microvilli-like structures are associated with the internalization of virulent capsulated Neisseria meningitidis into vascular endothelial cells. J. Cell Sci. 2002, 115, 1231–1241. [Google Scholar]
- Coureuil, M.; Lécuyer, H.; Scott, M.G.; Boularan, C.; Enslen, H.; Soyer, M.; Mikaty, G.; Bourdoulous, S.; Nassif, X.; Marullo, S. Meningococcus Hijacks a β2-adrenoceptor/β-Arrestin pathway to cross brain microvasculature endothelium. Cell 2010, 143, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Mikaty, G.; Soyer, M.; Mairey, E.; Henry, N.; Dyer, D.; Forest, K.T.; Morand, P.; Guadagnini, S.; Prevost, M.C.; Nassif, X.; et al. Extracellular bacterial pathogen induces host cell surface reorganization to resist shear stress. PLoS Pathog. 2009, 5, e1000314. [Google Scholar] [CrossRef] [Green Version]
- Doulet, N.; Donnadieu, E.; Laran-Chich, M.P.; Niedergang, F.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Neisseria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J. Cell Biol. 2006, 173, 627–637. [Google Scholar] [CrossRef]
- Slanina, H.; Mündlein, S.; Hebling, S.; Schubert-Unkmeir, A. Role of epidermal growth factor receptor signaling in the interaction of Neisseria meningitidis with endothelial cells. Infect. Immun. 2014, 82, 1243–1255. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, I.; Eugène, E.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Activation of ErbB2 receptor tyrosine kinase supports invasion of endothelial cells by Neisseria meningitidis. J. Cell Biol. 2001, 155, 133–143. [Google Scholar] [CrossRef] [Green Version]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Déry, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, O.; Heppel, N.; Jägerhuber, R.; Kim, K.S.; Frosch, M.; Eigenthaler, M.; Schubert-Unkmeir, A. Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: Role of MAP- and tyrosine kinases in invasion and inflammatory cytokine release. Cell. Microbiol. 2004, 6, 1153–1166. [Google Scholar] [CrossRef]
- Lambotin, M.; Hoffmann, I.; Laran-Chich, M.P.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Invasion of endothelial cells by Neisseria meningitidis requires cortactin recruitment by a phosphoinositide-3-kinase/Rac1 signalling pathway triggered by the lipo-oligosaccharide. J. Cell Sci. 2005, 118, 3805–3816. [Google Scholar] [CrossRef] [Green Version]
- Nikulin, J.; Panzner, U.; Frosch, M.; Schubert-Unkmeir, A. Intracellular survival and replication of Neisseria meningitidis in human brain microvascular endothelial cells. Int. J. Med Microbiol. 2006, 296, 553–558. [Google Scholar] [CrossRef]
- Coureuil, M.; Mikaty, G.; Miller, F.; Lecuyer, H.; Bernard, C.; Bourdoulous, S.; Dumenil, G.; Mege, R.M.; Weksler, B.B.; Romero, I.A.; et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science 2009, 325, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Schubert-Unkmeir, A.; Konrad, C.; Slanina, H.; Czapek, F.; Hebling, S.; Frosch, M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: A role for MMP-8. PLoS Pathog. 2010, 6, e1000874. [Google Scholar] [CrossRef] [Green Version]
- Schubert-Unkmeir, A.; Sokolova, O.; Panzner, U.; Eigenthaler, M.; Frosch, M. Gene expression pattern in human brain endothelial cells in response to Neisseria meningitidis. Infect. Immun. 2007, 75, 899–914. [Google Scholar] [CrossRef] [Green Version]
- Mairey, E.; Genovesio, A.; Donnadieu, E.; Bernard, C.; Jaubert, F.; Pinard, E.; Seylaz, J.; Olivo-Marin, J.C.; Nassif, X.; Dumenil, G. Cerebral microcirculation shear stress levels determine Neisseria meningitidis attachment sites along the blood-brain barrier. J. Exp. Med. 2006, 203, 1939–1950. [Google Scholar] [CrossRef] [Green Version]
- Ká, E.; Jiménez-Munguía, I.; Majerová, P.; Tkáčová, Z.; Bhide, K.; Mertinková, P.; Pulzová, L.; Kováč, A.; Bhide, M. Deciphering the Interactome of Neisseria meningitidis With Human Brain Microvascular Endothelial Cells. Front. Microbiol. 2018, 9, 2294. [Google Scholar] [CrossRef] [Green Version]
- Hardy, S.J.; Christodoulides, M.; Weller, R.O.; Heckels, J.E. Interactions of Neisseria meningitidis with cells of the human meninges. Mol. Microbiol. 2000, 36, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Unkmeir, A.; Latsch, K.; Dietrich, G.; Wintermeyer, E.; Schinke, B.; Schwender, S.; Kim, K.S.; Eigenthaler, M.; Frosch, M. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 2002, 46, 933–946. [Google Scholar] [CrossRef]
- Bradley, C.J.; Griffiths, N.J.; Rowe, H.A.; Heyderman, R.S.; Virji, M. Critical determinants of the interactions of capsule-expressing Neisseria meningitidis with host cells: The role of receptor density in increased cellular targeting via the outer membrane Opa proteins. Cell. Microbiol. 2005, 7, 1490–1503. [Google Scholar] [CrossRef]
- Schubert-Unkmeir, A. Molecular mechanisms involved in the interaction of Neisseria meningitidis with cells of the human blood–cerebrospinal fluid barrier. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef] [Green Version]
- Virji, M.; Makepeace, K.; Ferguson, D.J.; Watt, S.M. Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic neisseriae. Mol. Microbiol. 1996, 22, 941–950. [Google Scholar] [CrossRef]
- Virji, M.; Makepeace, K.; Moxon, E.R. Distinct mechanisms of interactions of Opc-expressing meningococci at apical and basolateral surfaces of human endothelial cells; the role of integrins in apical interactions. Mol. Microbiol. 1994, 14, 173–184. [Google Scholar] [CrossRef]
- Sa, E.C.C.; Griffiths, N.J.; Virji, M. Neisseria meningitidis Opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog. 2010, 6, e1000911. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef]
- Slanina, H.; König, A.; Hebling, S.; Hauck, C.R.; Frosch, M.; Schubert-Unkmeir, A. Entry of Neisseria meningitidis into mammalian cells requires the Src family protein tyrosine kinases. Infect. Immun. 2010, 78, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Slanina, H.; Hebling, S.; Hauck, C.R.; Schubert-Unkmeir, A. Cell invasion by Neisseria meningitidis requires a functional interplay between the focal adhesion kinase, Src and cortactin. PLoS ONE 2012, 7, e39613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sa, E.C.C.; Griffiths, N.J.; Murillo, I.; Virji, M. Neisseria meningitidis Opc invasin binds to the cytoskeletal protein alpha-actinin. Cell. Microbiol. 2009, 11, 389–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, A.; Reinhardt, R.; Sarkari, J.; Caugant, D.A.; Achtman, M. Allelic polymorphism and site-specific recombination in the opc locus of Neisseria meningitidis. Mol. Microbiol. 1996, 19, 841–856. [Google Scholar] [CrossRef]
- Kriz, P.; Vlckova, J.; Bobak, M. Targeted vaccination with meningococcal polysaccharide vaccine in one district of the Czech Republic. Epidemiol. Infect. 1995, 115, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whalen, C.M.; Hockin, J.C.; Ryan, A.; Ashton, F. The changing epidemiology of invasive meningococcal disease in Canada, 1985 through 1992. Emergence of a virulent clone of Neisseria meningitidis. JAMA 1995, 273, 390–394. [Google Scholar] [CrossRef]
- Oosthuysen, W.F.; Mueller, T.; Dittrich, M.T.; Schubert-Unkmeir, A. Neisseria meningitidis causes cell cycle arrest of human brain microvascular endothelial cells at S phase via p21 and cyclin G2. Cell. Microbiol. 2016, 18, 46–65. [Google Scholar] [CrossRef]
- Von Papen, M.; Oosthuysen, W.F.; Becam, J.; Claus, H.; Schubert-Unkmeir, A. Disease and Carrier Isolates of Neisseria meningitidis Cause G1 Cell Cycle Arrest in Human Epithelial Cells. Infect. Immun. 2016, 84, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Simonis, A.; Hebling, S.; Gulbins, E.; Schneider-Schaulies, S.; Schubert-Unkmeir, A. Differential activation of acid sphingomyelinase and ceramide release determines invasiveness of Neisseria meningitidis into brain endothelial cells. PLoS Pathog. 2014, 10, e1004160. [Google Scholar] [CrossRef] [Green Version]
- Pron, B.; Taha, M.K.; Rambaud, C.; Fournet, J.C.; Pattey, N.; Monnet, J.P.; Musilek, M.; Beretti, J.L.; Nassif, X. Interaction of Neisseria maningitidis with the components of the blood-brain barrier correlates with an increased expression of PilC. J. Infect. Dis. 1997, 176, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Nassif, X.; Bourdoulous, S.; Eugène, E.; Couraud, P.O. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 2002, 10, 227–232. [Google Scholar] [CrossRef]
- Guarner, J.; Greer, P.W.; Whitney, A.; Shieh, W.J.; Fischer, M.; White, E.H.; Carlone, G.M.; Stephens, D.S.; Popovic, T.; Zaki, S.R. Pathogenesis and diagnosis of human meningococcal disease using immunohistochemical and PCR assays. Am. J. Clin. Pathol. 2004, 122, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Woehrl, B.; Linn, J.; Lummel, N.; Pfefferkorn, T.; Koedel, U.; Pfister, H.W.; Klein, M. Pneumococcal meningitis-associated pyogenic ventriculitis. J. Infect. 2015, 70, 311–314. [Google Scholar] [CrossRef]
- Schwerk, C.; Papandreou, T.; Schuhmann, D.; Nickol, L.; Borkowski, J.; Steinmann, U.; Quednau, N.; Stump, C.; Weiss, C.; Berger, J.; et al. Polar Invasion and Translocation of Neisseria meningitidis and Streptococcus suis in a Novel Human Model of the Blood-Cerebrospinal Fluid Barrier. PLoS ONE 2012, 7, e30069. [Google Scholar] [CrossRef] [PubMed]
- Dinner, S.; Borkowski, J.; Stump-Guthier, C.; Ishikawa, H.; Tenenbaum, T.; Schroten, H.; Schwerk, C. A Choroid Plexus Epithelial Cell-based Model of the Human Blood-Cerebrospinal Fluid Barrier to Study Bacterial Infection from the Basolateral Side. J. Vis. Exp. Jove 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
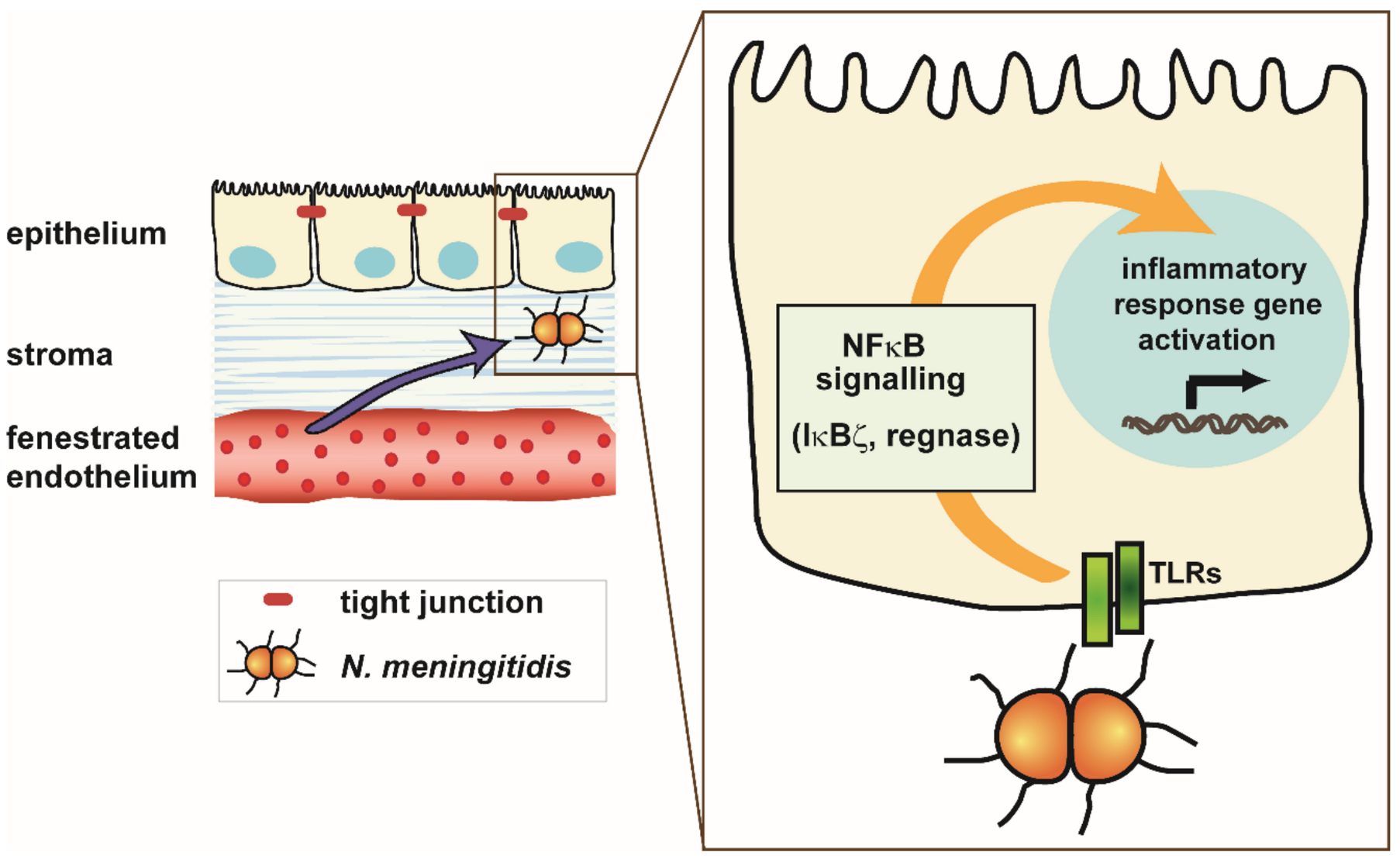
- Borkowski, J.; Li, L.; Steinmann, U.; Quednau, N.; Stump-Guthier, C.; Weiss, C.; Findeisen, P.; Gretz, N.; Ishikawa, H.; Tenenbaum, T.; et al. Neisseria meningitidis elicits a pro-inflammatory response involving IκBζ in a human blood-cerebrospinal fluid barrier model. J. Neuroinflamm. 2014, 11, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.W.; Gasque, P. How does the brain limit the severity of inflammation and tissue injury during bacterial meningitis? J. Neuropathol. Exp. Neurol. 2013, 72, 370–385. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, U.; Borkowski, J.; Wolburg, H.; Schröppel, B.; Findeisen, P.; Weiss, C.; Ishikawa, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Transmigration of polymorphnuclear neutrophils and monocytes through the human blood-cerebrospinal fluid barrier after bacterial infection in vitro. J. Neuroinflamm. 2013, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Schwerk, C.; Adam, R.; Borkowski, J.; Schneider, H.; Klenk, M.; Zink, S.; Quednau, N.; Schmidt, N.; Stump, C.; Sagar, A.; et al. In vitro transcriptome analysis of porcine choroid plexus epithelial cells in response to Streptococcus suis: Release of pro-inflammatory cytokines and chemokines. Microbes Infect. Inst. Pasteur 2011, 13, 953–962. [Google Scholar] [CrossRef]
- Toussi, D.N.; Wetzler, L.M.; Liu, X.; Massari, P. Neisseriae internalization by epithelial cells is enhanced by TLR2 stimulation. Microbes Infect. 2016, 18, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Massari, P.; Visintin, A.; Gunawardana, J.; Halmen, K.A.; King, C.A.; Golenbock, D.T.; Wetzler, L.M. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J. Immunol. 2006, 176, 2373–2380. [Google Scholar] [CrossRef] [Green Version]
- Massari, P.; Henneke, P.; Ho, Y.; Latz, E.; Golenbock, D.T.; Wetzler, L.M. Cutting edge: Immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 2002, 168, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Massari, P.; Ram, S.; Macleod, H.; Wetzler, L.M. The role of porins in neisserial pathogenesis and immunity. Trends Microbiol. 2003, 11, 87–93. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamazaki, S.; Uematsu, S.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Kuwata, H.; Takeuchi, O.; Takeshige, K.; et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature 2004, 430, 218–222. [Google Scholar] [CrossRef]
- Zughaier, S.M. Neisseria meningitidis capsular polysaccharides induce inflammatory responses via TLR2 and TLR4-MD-2. J. Leukoc. Biol. 2011, 89, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laflamme, N.; Rivest, S. Toll-like receptor 4: The missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001, 15, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laflamme, N.; Soucy, G.; Rivest, S. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J. Neurochem. 2001, 79, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Feurer, D.J.; Weller, R.O. Barrier functions of the leptomeninges: A study of normal meninges and meningiomas in tissue culture. Neuropathol. Appl. Neurobiol. 1991, 17, 391–405. [Google Scholar] [CrossRef]
- Humphries, H.E.; Triantafilou, M.; Makepeace, B.L.; Heckels, J.E.; Triantafilou, K.; Christodoulides, M. Activation of human meningeal cells is modulated by lipopolysaccharide (LPS) and non-LPS components of Neisseria meningitidis and is independent of Toll-like receptor (TLR)4 and TLR2 signalling. Cell. Microbiol. 2005, 7, 415–430. [Google Scholar] [CrossRef]
- Wells, D.B.; Tighe, P.J.; Wooldridge, K.G.; Robinson, K.; Ala’ Aldeen, D.A. Differential gene expression during meningeal-meningococcal interaction: Evidence for self-defense and early release of cytokines and chemokines. Infect. Immun. 2001, 69, 2718–2722. [Google Scholar] [CrossRef] [Green Version]
- Fowler, M.I.; Weller, R.O.; Heckels, J.E.; Christodoulides, M. Different meningitis-causing bacteria induce distinct inflammatory responses on interaction with cells of the human meninges. Cell. Microbiol. 2004, 6, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Taraktsoglou, M.; Rowe, K.S.; Wooldridge, K.G.; Ala’Aldeen, D.A. Secreted proteins from Neisseria meningitidis mediate differential human gene expression and immune activation. Cell. Microbiol. 2004, 6, 927–938. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Nasopharyngeal Epithelium | Peripheral and Cerebral Endothelium |
|---|---|
| • Formation of cortical plaques [35,87,88] • Recruitment of ezrin [35,87,88] • Accumulation of actin [22,87] • Recruitment of adhesion molecules [35,87,88] • Recruitment of membrane receptors [35,87,88] • Recruitment of β2AR [76,88] | |
| Recruitment of the polarity complex (Par3/Par6) beneath attached colonies colocalizing with cortical plaques [99] |
| Recruitment of junctional components (AJ: VE-cadherin, p120 catenin, and β-catenin; and TJ: ZO1, ZO2, and claudin-5) under the attached microcolonies [99] |
| Formation of gaps between the cells promoting paracellular barrier crossing [99] | |
| Activation of β-arrestin pathway upon adhesion [88] |
| β-arrestin signaling pathway is essential for the formation of cortical plaques [88] |
| Recruitment of Src kinase below the attached colonies, Src involvement in formation of cortical plaques, and actin polymerization [76] |
| protrusions stabilize the colonies and protect them against blood flow generated shear stress [90] |
| Activation of the β-arrestin pathway is PilV (and PilE) dependent; Ezrin recruitment, cortical plaque, and shear stress resistance are PilV dependent [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkowski, J.; Schroten, H.; Schwerk, C. Interactions and Signal Transduction Pathways Involved during Central Nervous System Entry by Neisseria meningitidis across the Blood–Brain Barriers. Int. J. Mol. Sci. 2020, 21, 8788. https://doi.org/10.3390/ijms21228788
Borkowski J, Schroten H, Schwerk C. Interactions and Signal Transduction Pathways Involved during Central Nervous System Entry by Neisseria meningitidis across the Blood–Brain Barriers. International Journal of Molecular Sciences. 2020; 21(22):8788. https://doi.org/10.3390/ijms21228788
Chicago/Turabian StyleBorkowski, Julia, Horst Schroten, and Christian Schwerk. 2020. "Interactions and Signal Transduction Pathways Involved during Central Nervous System Entry by Neisseria meningitidis across the Blood–Brain Barriers" International Journal of Molecular Sciences 21, no. 22: 8788. https://doi.org/10.3390/ijms21228788
APA StyleBorkowski, J., Schroten, H., & Schwerk, C. (2020). Interactions and Signal Transduction Pathways Involved during Central Nervous System Entry by Neisseria meningitidis across the Blood–Brain Barriers. International Journal of Molecular Sciences, 21(22), 8788. https://doi.org/10.3390/ijms21228788