2.6. Analysis of Phage Genomes
DNA was isolated from purified virions of vB_Sen-TO17 and vB_Sen-E22 and subjected to sequencing, as described in
Section 4.14. Annotations of vB_Sen-TO17 and vB_Sen-E22 genomes are presented in
Tables S1 and S2, respectively.
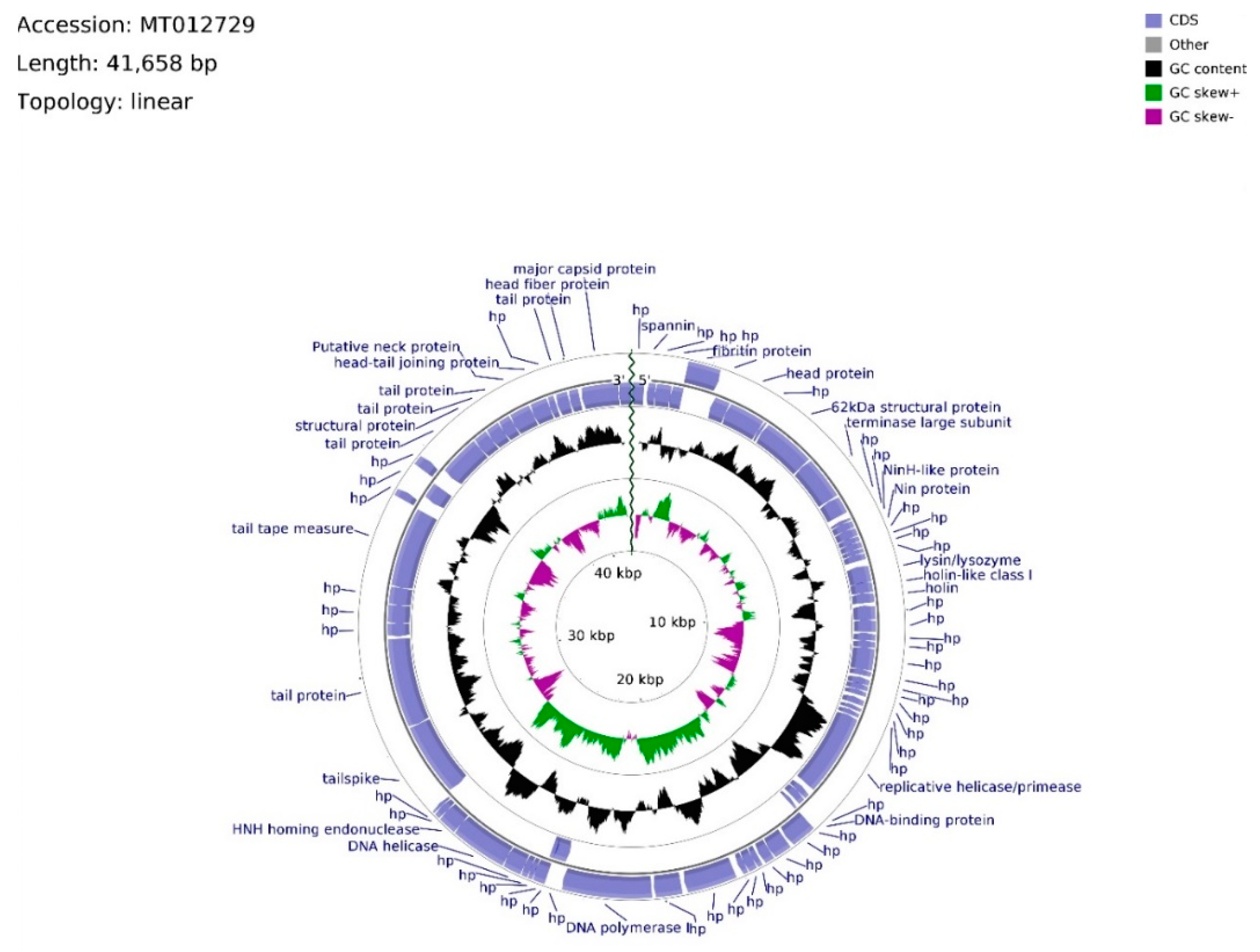
The genome of phage vB_Sen-TO17 (whole sequence deposited in GenBank; accession no. MT012729) consists of 41,658 bps, arranged in a linear topology with an overall GC content of 50.78% (
Figure 5). We identified open reading frames (ORFs) putatively coding for 75 proteins, of which 27 were reported previously. The remaining 48 ORFs are described as hypothetical.
Twenty ORFs are spread out on the leading strand, whereas the majority (55) of ORFs are located on the complementary strand. ATG codon predominates among start codons (70 cases), whereas GTG and TTG occur 3 and 2 times, respectively. ORFs initiating with GTG and TTG codons, with the exception of vB_SenTO17_45 (696 bp), have sequence spans ≤ 144 bp, where the average length of sequence in the vB_Sen-TO17 genome is 535 bp. ATG codon is utilized in every ORF with an assigned function. The frequency of observed stop codons is set out as follows: TAA—40, TGA—28, TAG—7. ORFs were divided into four functional groups due to the assigned functions of their putative products: Morphogenesis (15), DNA replication (4), lysis (3), and DNA packing (2). The total number of ORFs in functional groups was excessive due to the domain determination of hypothetical proteins. In consequence, putatively protein-encoding vB_SenTO17_52, bearing the HNH endonuclease domain sequence, was classified within the DNA replication functional group, joined with vB_SenTO17_34, vB_SenTO17_36, vB_SenTO17_46, and vB_SenTO17_53, encoding helicase/primase, DNA-binding protein, DNA polymerase I, and DNA helicase (which start codon overlaps vB_SenTO17_52 stop codon), respectively. DNA replication genes are spread within the 12,966–25,568 bps span. The majority of ORF coding proteins putatively engaged in morphogenesis are located downstream, within the 26,354–41,287 bps region, with the exception of genes encoding fibritin (vB_SenTO17_06), putative head protein (vB_SenTO17_07), and 62 kDa structural protein (vB_SenTO17_09), located at the 2282–5401 bps region. Numerous possible transcription promoters were registered on the complementary strand in the morphogenesis-related region, in relation to the whole genome. The morphogenesis group consists mostly of genes coding for proteins involved in tail assembly (vB_SenTO17_07, vB_SenTO17_57, vB_SenTO17_58, vB_SenTO17_62, vB_SenTO17_66, vB_SenTO17_68, vB_SenTO17_69, vB_SenTO17_73), head (vB_SenTO17_74, vB_SenTO17_75), head–tail joining proteins (vB_SenTO17_70, vB_SenTO17_71), and structural proteins (vB_SenTO17_09, vB_SenTO17_67). Coding DNA Sequences (CDSs) corresponding to lysis proteins are located upstream of the DNA replication span, and occupy positions at 8694–9739 bps. They include ORFs for lysin/lysozyme (vB_SenTO17_19), putative holin-like class I (vB_SenTO17_20), and putative holin (vB_SenTO17_21). Nevertheless, based on nucleotide sequence analysis using the PHACTS algorithm, phage lifestyle was non-confidently classified as temperate. DNA packing CDSs are located within the 5418–6641 bps region, and the 25,565–26,062 bps span consists of genes putatively encoding the terminase large subunit (vB_SenTO17_10) and the HNH homing endonuclease (vB_SenTO17_54). Li’s method analysis suggested that phage vB_Sen-TO17 genome is packaged according to the PAC system. Following ORFs: vB_SenTO17_54, vB_SenTO17_53, and vB_SenTO17_46 are the only sequences located on the leading strand which may code for proteins with the reported function.
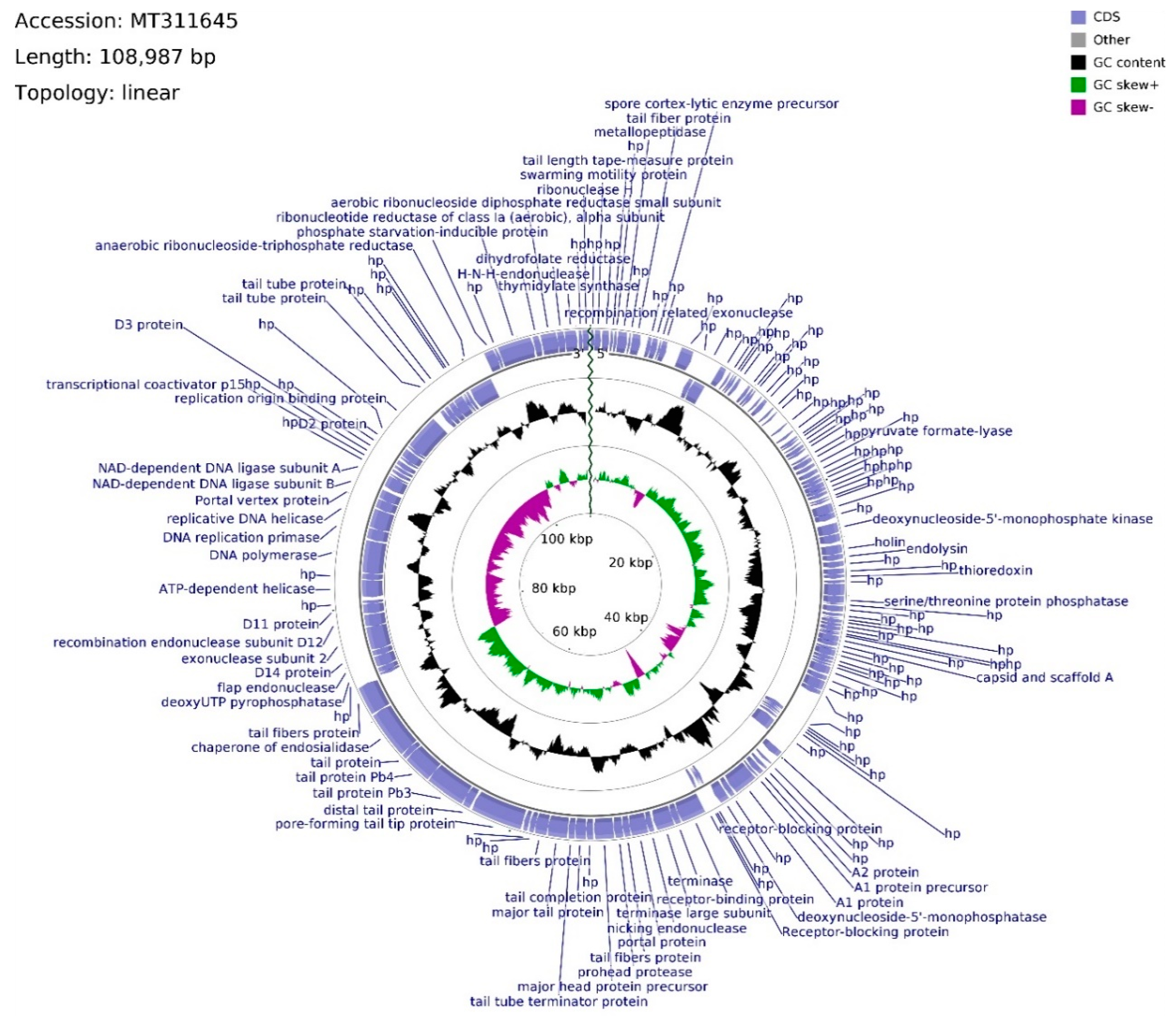
The genome of phage vB_Sen-E22 (whole sequence deposited in GenBank; accession no. MT311645) consists of 108,987 bp, with overall GC content of 39.21% and linear topology (
Figure 6). Determination of ORFs distinguished 158 putative protein-coding genes where 114 were located on the leading strand and 44 were located on the complementary strand. The frequency of start codons is set out as follows: ATG—147, GTG—8, TTG—3. Among termination codons, nucleotide triplet frequencies were set as follows: TAA—124, TGA—25, TAG—8. The functions of 67 ORFs were assigned, whereas 91 remain hypothetical. ORFs with assigned functions were divided into four functional groups: Morphogenesis (22), DNA packing (10), DNA replication (7), and lysis (2). Morphogenesis CDSs are concentrated inside the region of 46,010–74,545 bps, with nine sequences interspersed throughout the vB_Sen-E22 genome, mainly coding for tail-related proteins with the exception of head assembly proteins vB_SenE22_68 and vB_SenE22_131. CDSs engaged in tail protein assembly dominate this genome region (11 CDSs), whereas two head-related CDSs, coding for portal protein (vB_SenE22_100) and major head protein precursor (vB_SenE22_103), are also present there. Phage head putative genes are located within the 50,510–54,248 bps span which is intersected with the tail fibers protein putative gene (vB_SenE22_101) and the sequence putatively encoding prohead protease (vB_SenE22_102). DNA packing genes are spread across the vB_Sen-E22 genome, and they include genes encoding putative nucleases: Endonucleases (vB_SenE22_99, vB_SenE22_120, vB_SenE22_123, vB_SenE22_153), exonucleases (vB_SenE22_12, vB_SenE22_112), and ribonuclease H (vB_SenE22_158).
Apart from ORF for potential nicking endonuclease (vB_SenE22_99), two terminase subunit ORFs are situated on the leading strand which are preceded by CDSs of three receptor-blocking proteins. Using Li’s method, the DNA packaging of this phage can be suggested as operating by the COS mode. ORF for recombination-related exonuclease (vB_SenE22_12) is located upstream of the one for the hypothetical protein bearing PHB domain putatively engaged in phage decision between lytic and lysogenic growth. Within this domain, a Rho-independent terminator is located between vB_SenE22_12 and vB_SenE22_13 CDSs. The gap of the non-coding region encompasses 943 bps, whereas the average length of a gap between coding DNA sequences across the genome is equal to 93 bps. ORFs coding for proteins putatively involved in the process of DNA replication, located on the complementary strand, are assembled in a tile-like manner at the 80,720–90,335 bps region, with the putative replication origin binding protein ORF (vB_SenE22_141) situated upstream of the CDS conglomerate. vB_SenE22_141 (93,024–95,813 bp) overlaps with the vB_SenE22_140 hypothetical protein gene, bearing two transcription terminators starting at positions 117 bp and 183 bp inside the 234 bps long CDS. A transcription terminator can also be found within vB_SenE22_141 CDS, and downstream from the DNA ligase subunit B gene (vB_SenE22_132), which overlaps the A subunit ORF (vB_SenE22_133). The DNA replication tile is interlaced with the ORF encoding uncharacterized protein and the Portal vertex (vB_SenE22_131), belonging to the morphogenesis functional group. Between those sequences, there are ORFs for DNA helicase, DNA replication primase, and DNA polymerase, which are probably transcribed as two operons, as suggested by an overlap between start and stop codons with a 62 bps gap between vB_SenE22_129 and vB_SenE22_128. Based on the sequence analysis with PHACTS, this phage was non-confidently classified as lytic. Putative holin and endolysin CDSs (vB_SenE22_50 and vB_SenE22_51, respectively), representing genes coding for proteins involved in host cell lysis, overlap at positions 24,499–25,565 bps, shifting the probability of the lifestyle classification.
2.7. Phylogenetic Analyses
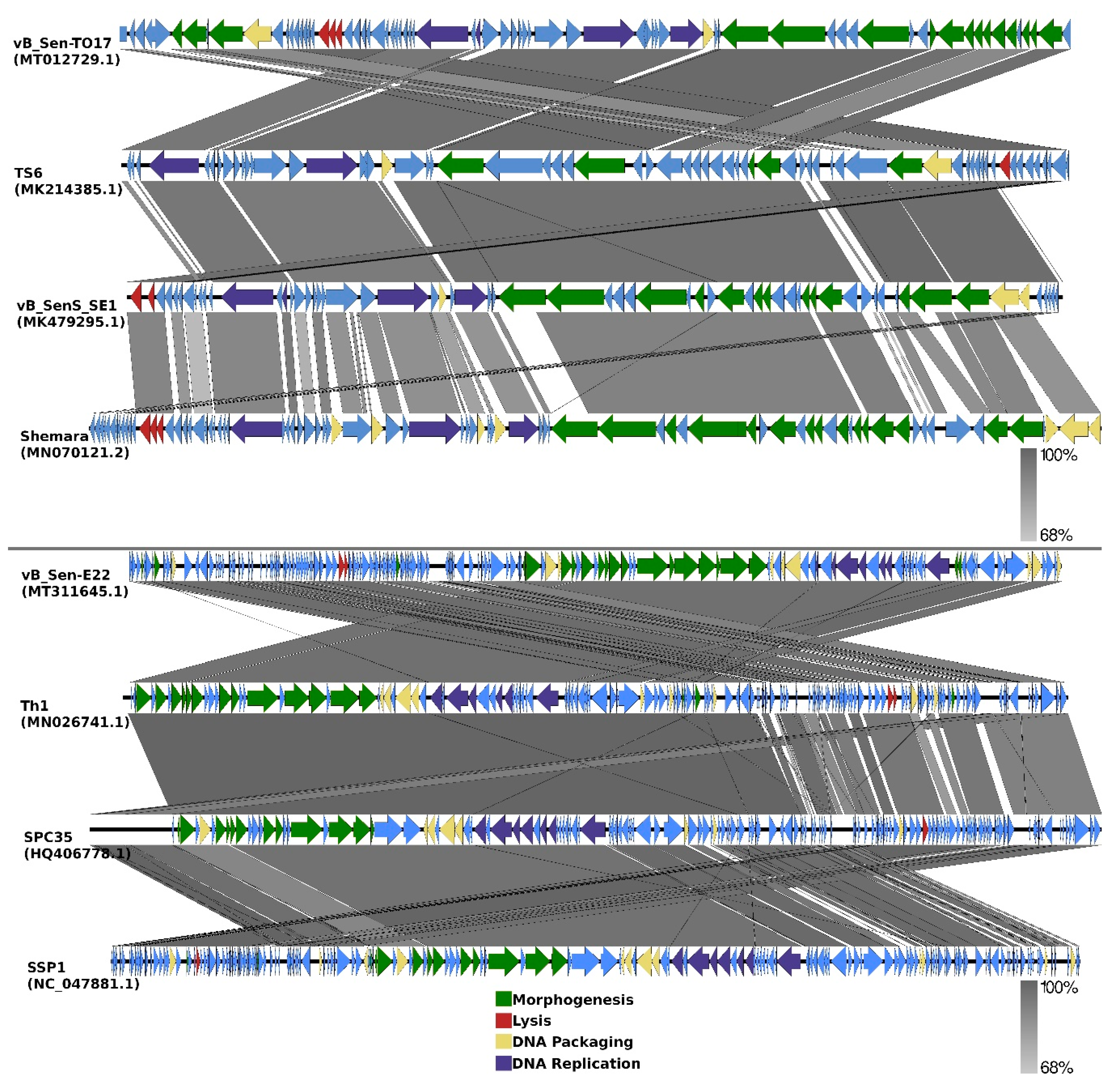
Comparisons of organizations of genomes of phages vB_Sen-TO17 and vB_Sen-E22 to genomes of the most related bacteriophages (according to DNA sequence similarities of whole genomes) are indicated as EasyFig in
Figure 7.
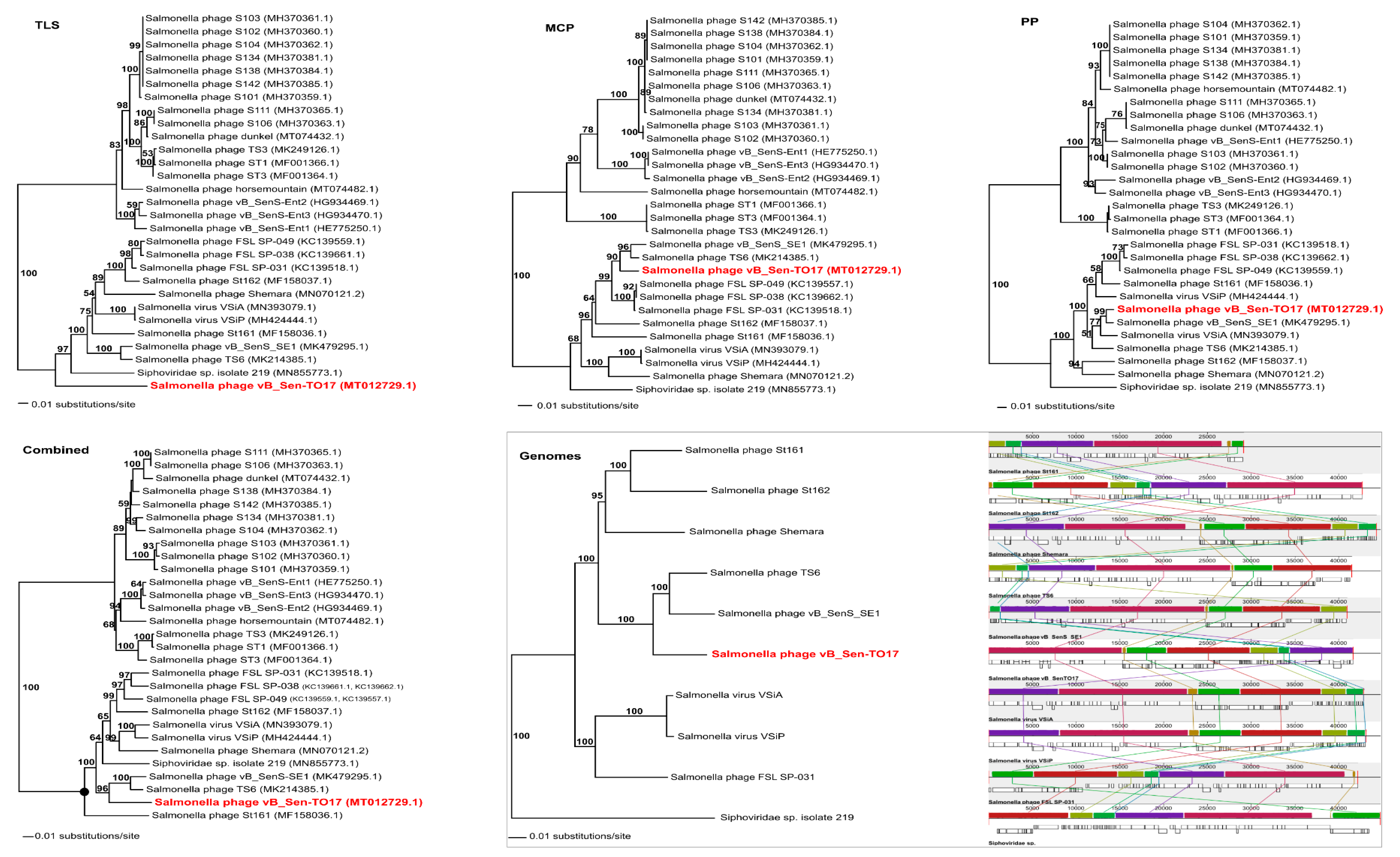
To analyse phylogenetic relationships between phage vB_Sen-TO17 and other viruses, we have compared the nucleotide sequences of the gene of the terminase large subunit and the nucleotide sequences of two additional markers such as genes encoding the portal protein and the major capsid protein of vB_Sen-TO17 with the respective sequences of other phages (
Figure 8). The use of only the large terminase subunit gene sequence was insufficient to show the actual phylogenetic position of vB_Sen-TO17. The analysis of the nucleotide sequence of the gene coding for portal protein indicated that phage vB_Sen-TO17 is a sister to phage vB_SenS_SE1 (MK479295.1) with high bootstrap support (BS = 99). On the other hand, as shown in
Figure 8, the analysis of the major capsid protein gene together with the combined analysis of three marker genes’ sequences (
TLS, PP, MCP) indicated a close relationship of vB_Sen-TO17 to phages vB_SenS_SE1 (MK479295.1) and TS6 (MK214385.1) with high bootstrap support of BS = 90 and BS = 96, respectively. Both phages belong to the family
Siphoviridae, genus
Cornellvirus. Sequence similarity searches of these phages demonstrated very high level of genome sequence identity with vB_Sen-TO17 (~96% and ~98%, respectively). The above results were confirmed by the whole-genome phylogenetic analysis and the whole-genome alignments constructed using the Mauve algorithm (see
Section 4.15 for details), which indicated a high level of homology between these genomes (
Figure 8). Nevertheless, one should note some differences in the trees topology obtained with the use of various methods presented above, for example in the position of Shemara phage (MN070121.2).
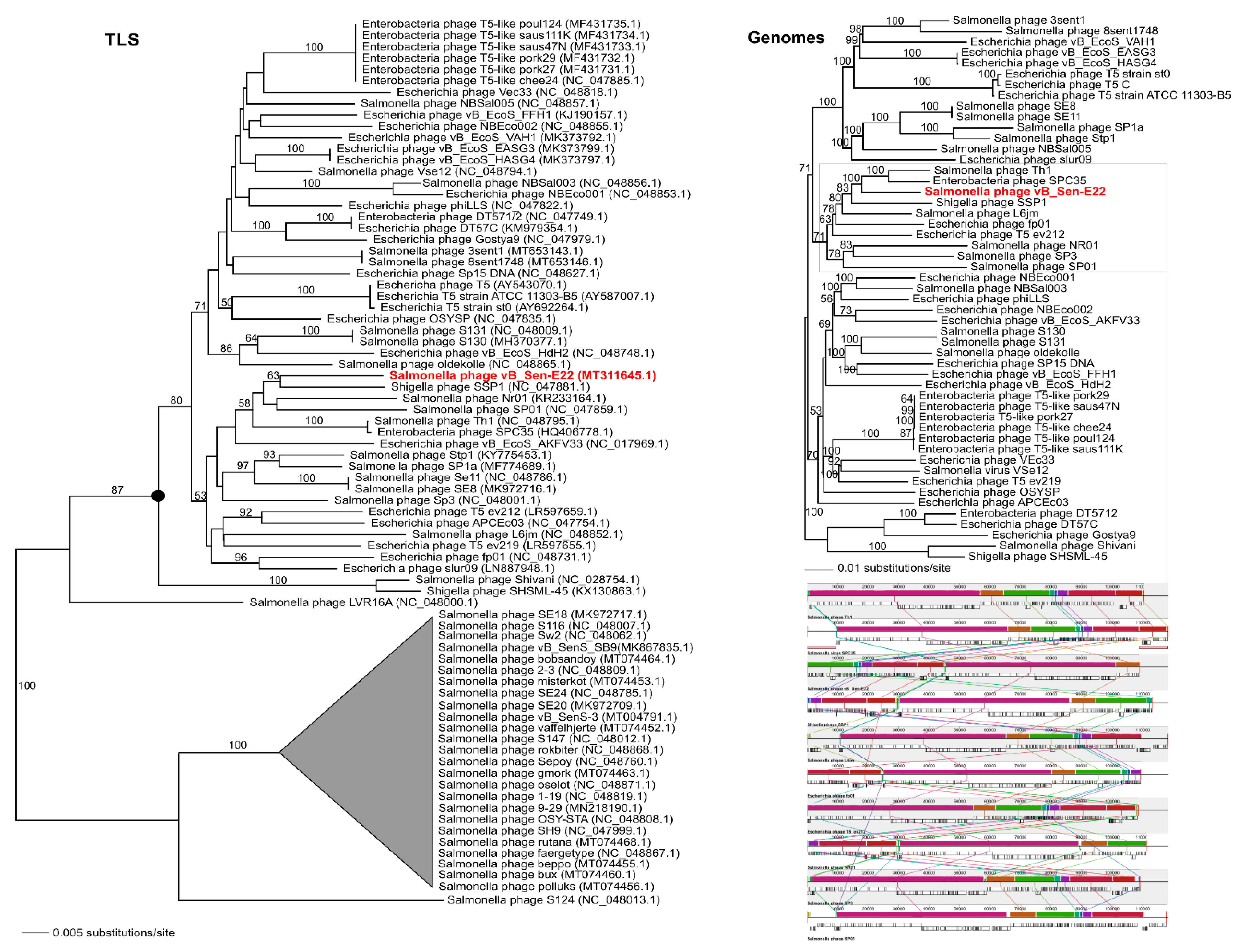
To analyse phylogenetic relationships between phage vB_Sen-E22 and other viruses, we compared the nucleotide sequence of the gene coding for the large terminase subunit of vB_Sen-E22 with the sequences of genes of the large terminase subunit of other phages. As shown in
Figure 9, the sequence of the large terminase subunit gene of vB_Sen-E22 indicates its relationship to
Shigella phage SSP1 (NC_047881.1) with low bootstrap support BS = 63. On the other hand, the sequence similarity searches revealed that these phages show a very high level of genome sequence identity (~97%). The whole genome sequence analysis indicated that the phage vB_Sen-E22 is a sister to the phages Th1 (NC_048795.1) and SPC35 (HQ406778.1), with the highest BS = 100 in both cases (
Figure 9). Phages SSP1, Th1, and SPC35 belong to the family
Demerecviridae, genus
Tequintavirus. Sequence similarity searches between Th1, SPC35, and phage vB_Sen-E22 demonstrated a very high level of identity (~97% when comparing vB_Sen-E22 with Th1, and ~96% when comparing vB_Sen-E22 with SPC35). The whole-genome alignments constructed using the Mauve algorithm (see
Section 4.15 for details) also revealed a high level of homology between these genomes (
Figure 9). Therefore, for vB_Sen-E22, the single marker gene phylogenetic analysis was insufficient and did not reflect the actual genetic position of this phage, probably due to mosaicism of phage genomes and a horizontal gene transfer. Nevertheless, the whole genome sequence phylogenetic analysis allowed us to obtain reliable results, leading to the proposal that vB_Sen-E22 belongs to the family
Demerecviridae, genus
Tequintavirus.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}