A Rapid Robust Method for Subgrouping Non-NF2 Meningiomas According to Genotype and Detection of Lower Levels of M2 Macrophages in AKT1 E17K Mutated Tumours

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Mutational Hotspots were Detected at the Predicted Frequencies
2.2. Correlation of Genomic Subgroup with Clinical Characteristics
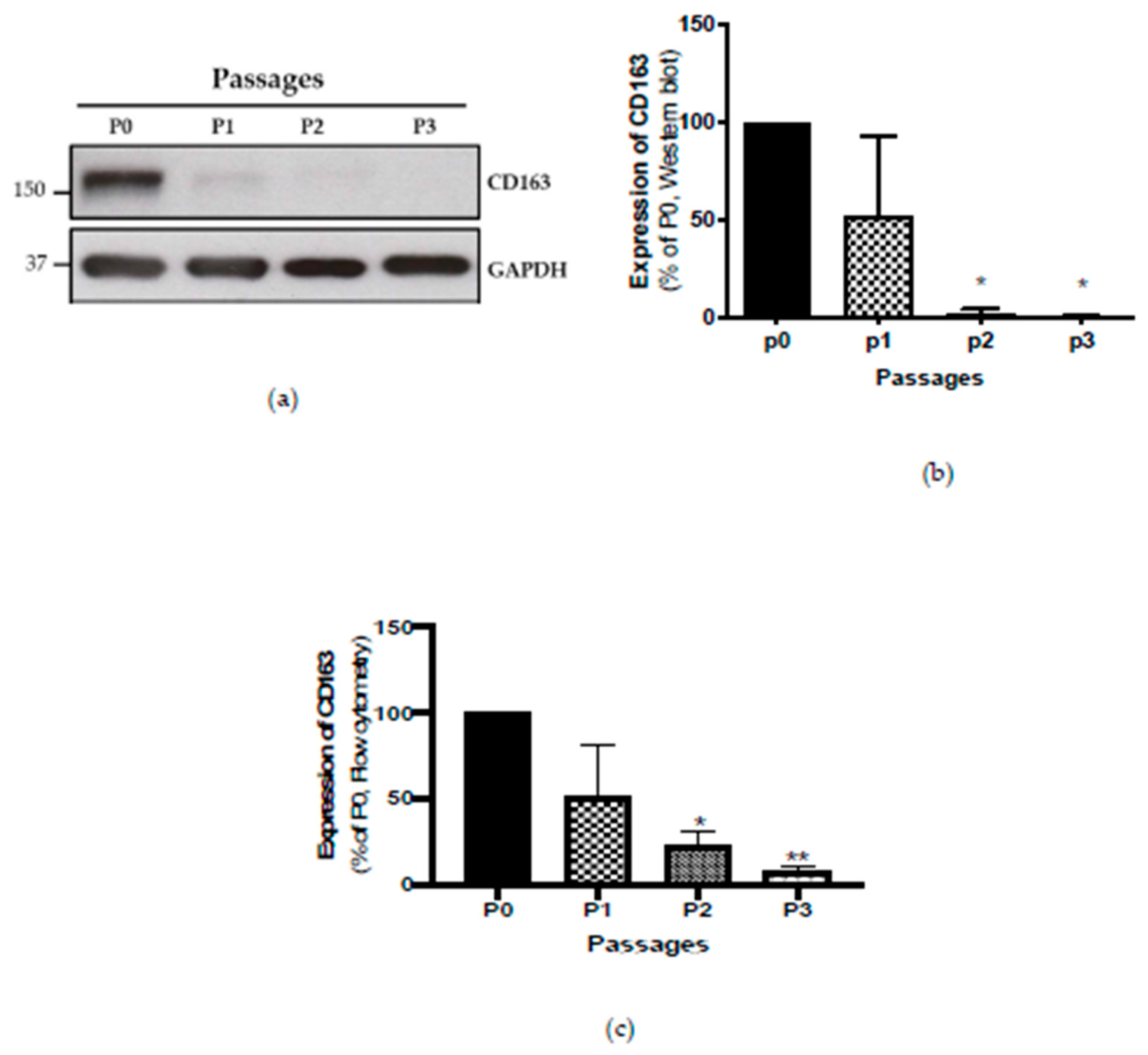
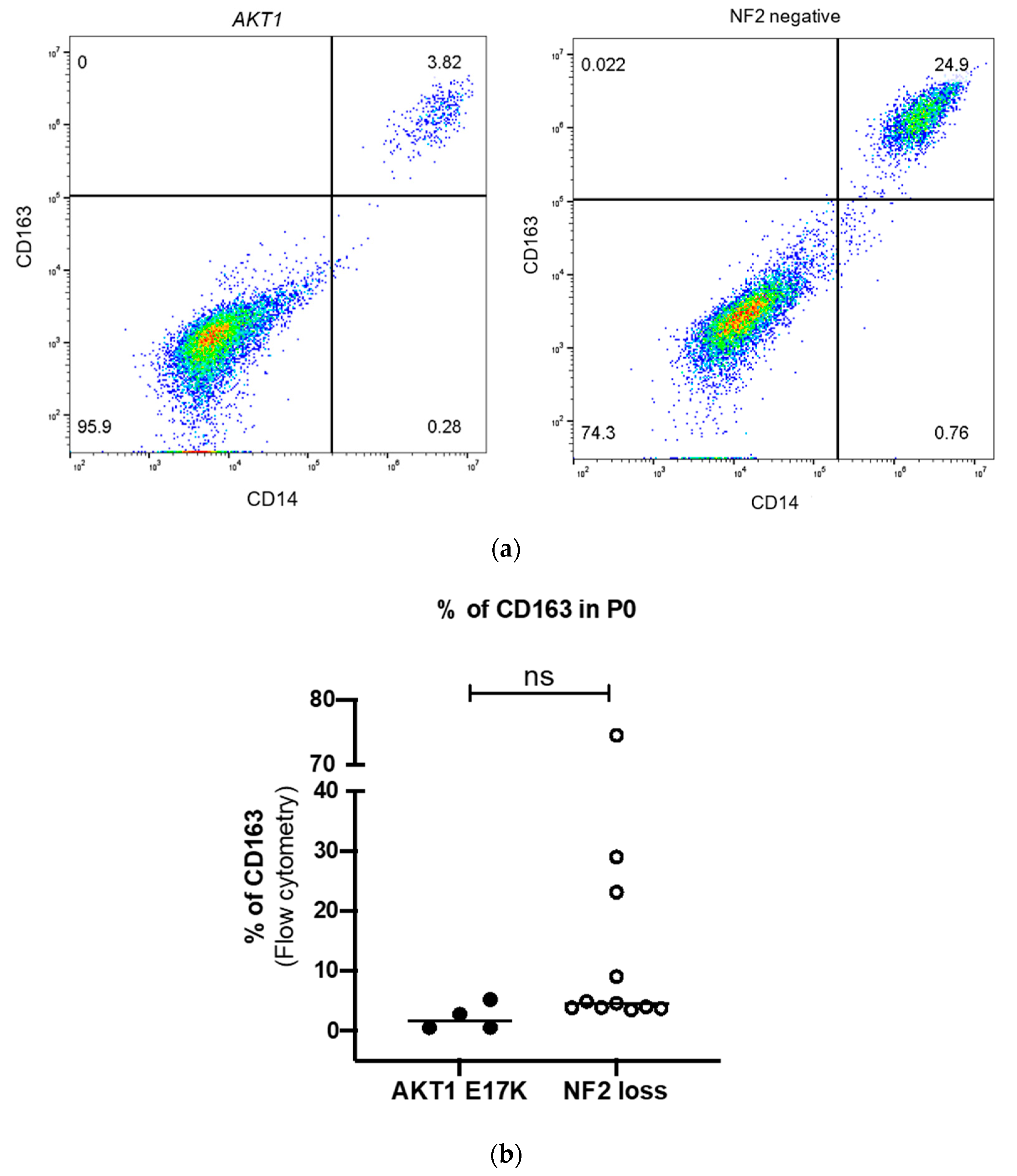
2.3. Detection of M2 Macrophages by Four-Colour Flow Cytometry
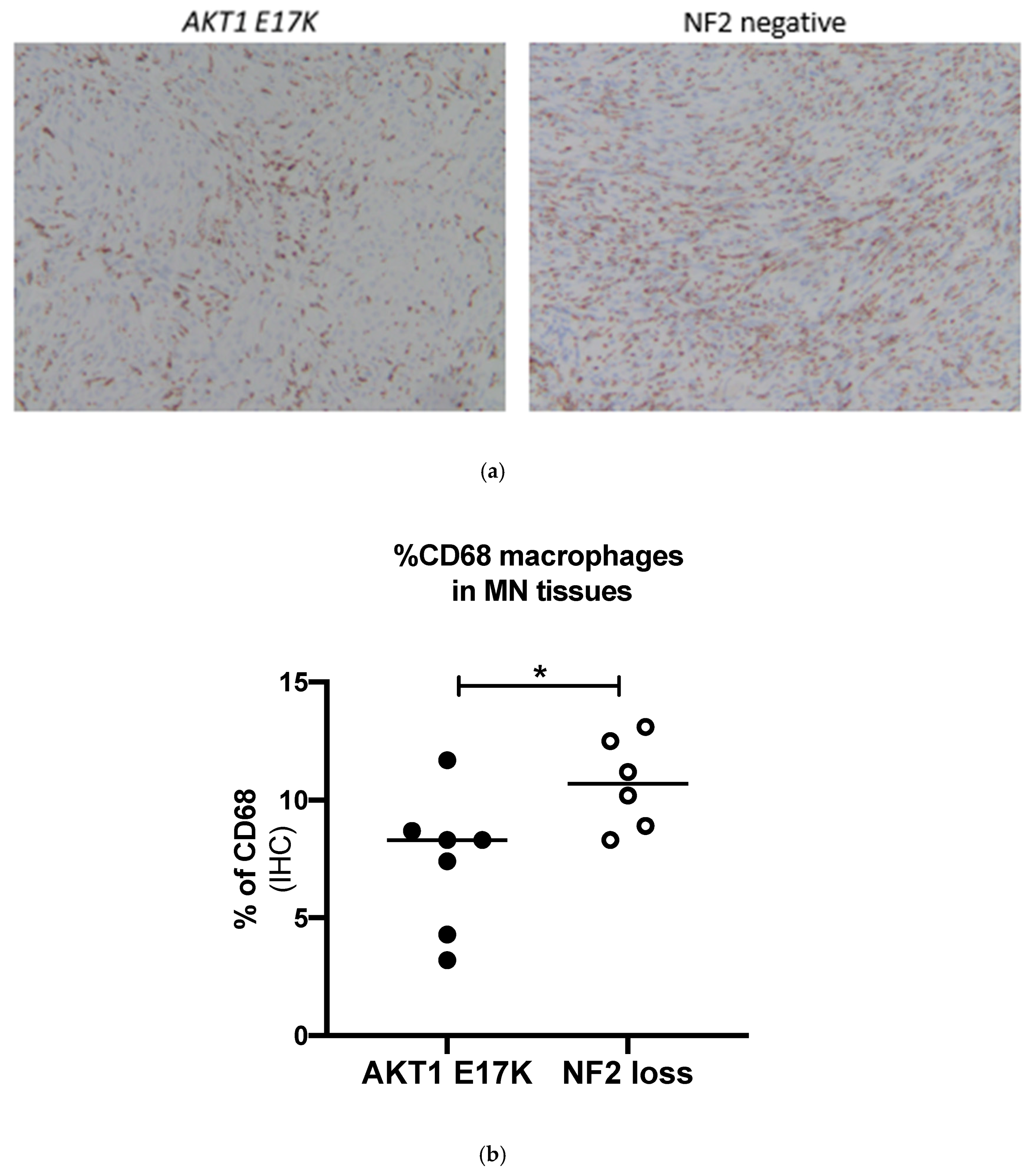
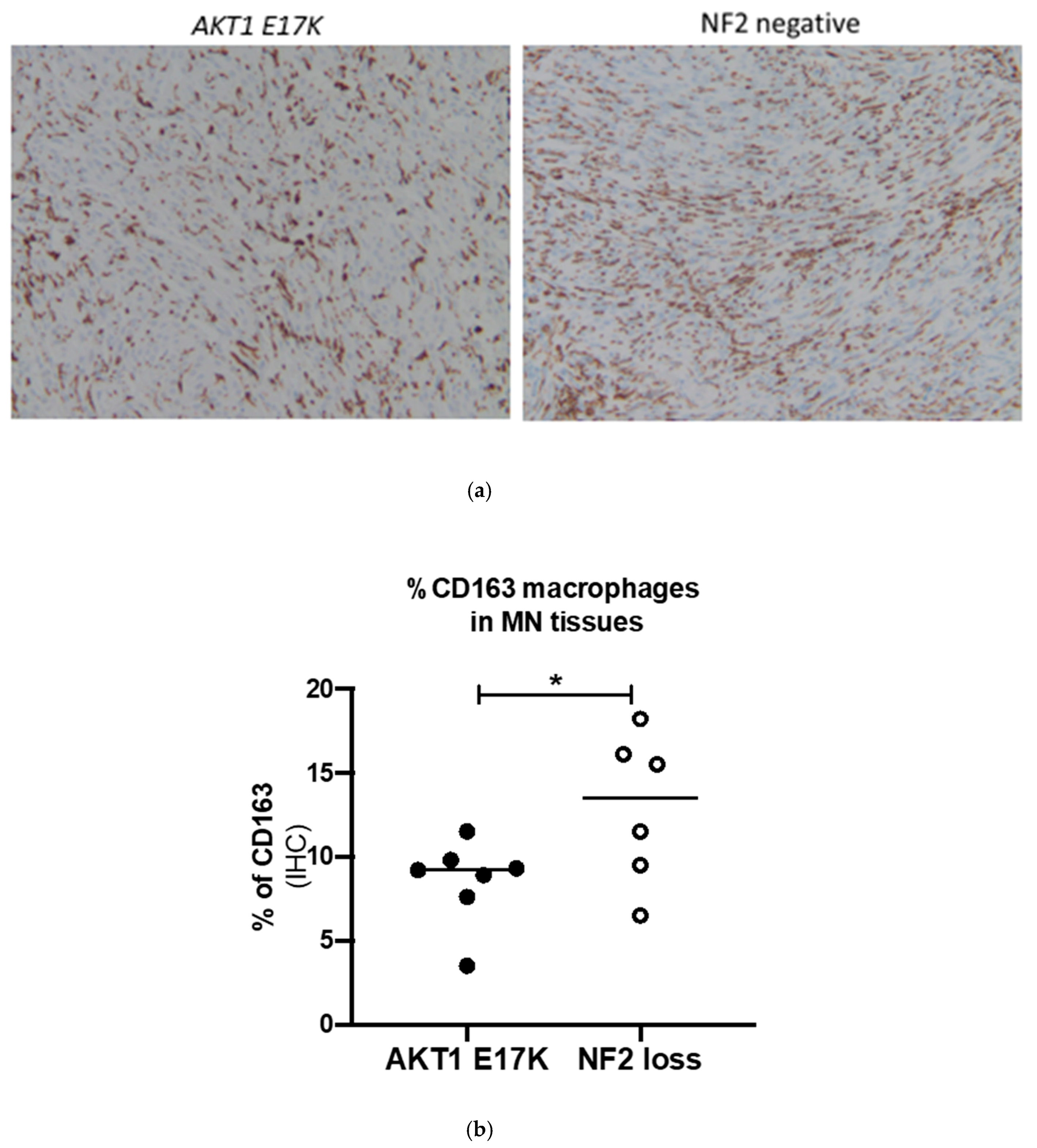
2.4. Confirmation of Macrophage Levels Using Immunohistochemistry
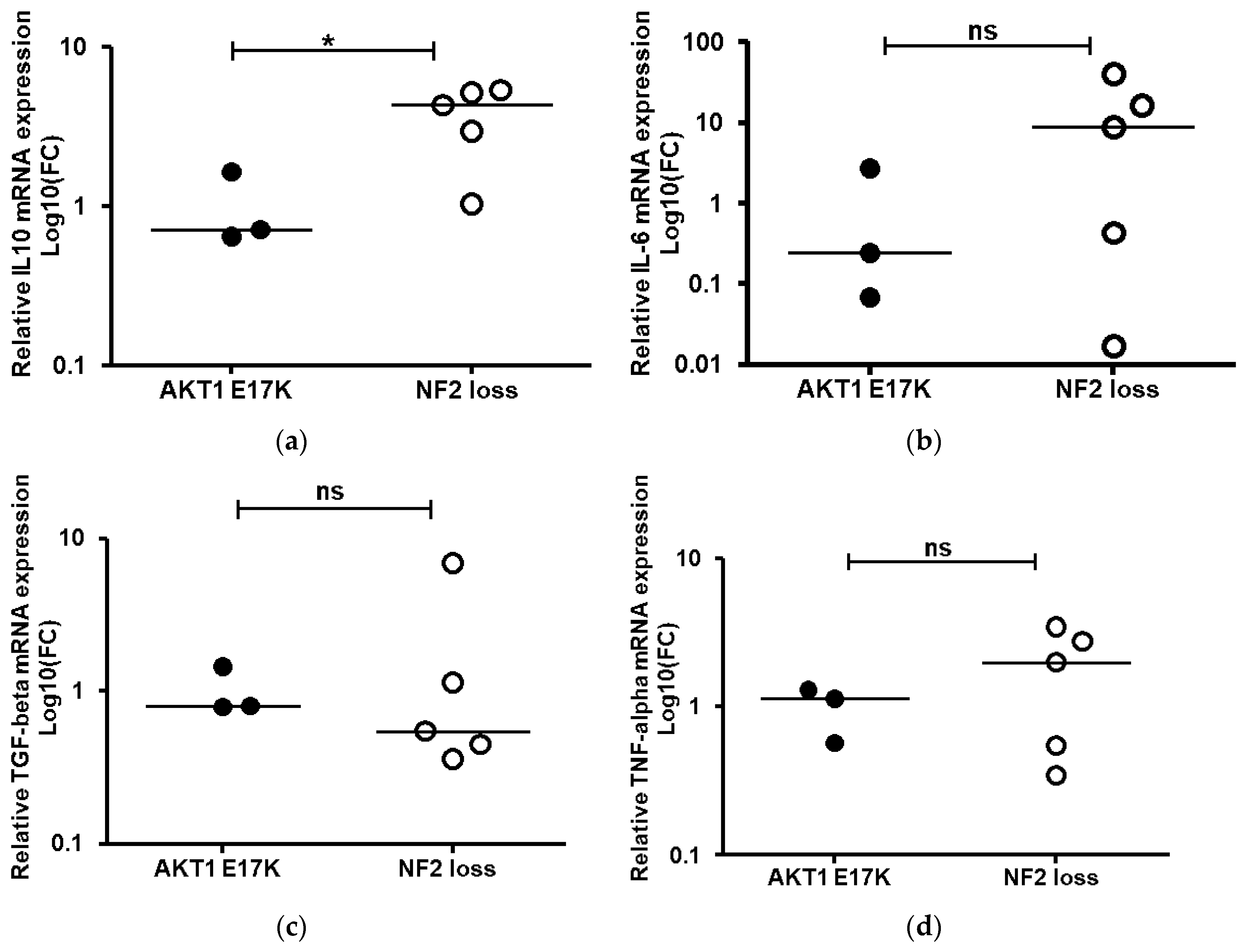
2.5. Assessment of Cytokine mRNA Expression Associated with Activated M2 Macrophages
3. Discussion
4. Methods and Materials
4.1. Clinical Samples
4.2. End Point Genotyping Method
4.3. Sanger Sequencing
4.4. Western Blotting
4.5. Isolation of Primary Meningioma Cells from Fresh Tissue
4.6. Flow Cytometry
4.7. Immunohistochemistry
4.8. Quantitative Real-Time PCR Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT1 | v-akt murine thymona viral oncogene |
| CD | Cluster of differentiation |
| GAPDH | glyceraldehyde 3-phosphate dehygrogenase |
| HLA | Human leukocyte antigen |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| KASP | Kompetitive allele specific PCR |
| KLF4 | Krupple like factor 4 |
| NF2 | Neurofibromatosis 2 tumour suppressor gene |
| PCR | Polymerase Chain Reaction |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| POLR2A | RNA Polymerase II Subunit A |
| SMARCB1 | SW1/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily B, member 1. |
| SMO | Smoothened, frizzled family receptor |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumour necrosis factor alpha |
| TRAF7 | Tumour necrosis factor receptor associated factor 7 |
| WHO | World Health Organisation |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Suppiah, S.; Nassiri, F.; Bi, W.L.; Dunn, I.F.; Hanemann, C.O.; Horbinski, C.M.; Hashizume, R.; James, C.D.; Mawrin, C.; Noushmehr, H.; et al. Molecular and translational advances in meningiomas. Neuro. Oncol. 2019, 21, i4–i17. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.T.; Ramachandran, S.; Lama, S.; Sutherland, G.R. Towards molecular classification of meningioma: Evolving treatment and diagnostic paradigms. World Neurosurg. 2018, 119, 366–373. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Galanis, E.; Butowski, N.; Chan, J.W.; Dunn, I.F.; Goldbrunner, R.; Herold-Mende, C.; Ippen, F.M.; Mawrin, C.; McDermott, M.W.; et al. Advances in multidisciplinary therapy for meningiomas. Neuro. Oncol. 2019, 21, i18–i31. [Google Scholar] [CrossRef] [Green Version]
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K.; et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Bi, W.L.; Zhang, M.; Wu, W.W.; Mei, Y.; Dunn, I.F. Meningioma genomics: Diagnostic, prognostic, and therapeutic applications. Front. Surg. 2016, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and Akt1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Yaniv, M. Chromatin remodeling: From transcription to cancer. Cancer Genet. 2014, 207, 352–357. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2017, 2, 1–4. [Google Scholar]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Santagata, S. BAP1 mutations in high-grade meningioma: Implications for patient care. Neuro-oncology 2017, 19, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro. Oncol. 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlSahlawi, A.; Aljelaify, R.; Magrashi, A.; AlSaeed, M.; Almutairi, A.; Alqubaishi, F.; Alturkistani, A.; AlObaid, A.; Abouelhoda, M.; AlMubarak, L.; et al. New insights into the genomic landscape of meningiomas identified FGFR3 in a subset of patients with favorable prognoses. Oncotarget 2019, 10, 5549–5559. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, S.; Nishihara, H.; Tanaka, S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016, 33, 237–247. [Google Scholar] [CrossRef]
- Smith, M.J. Germline and somatic mutations in meningiomas. Cancer Genet. 2015, 208, 107–114. [Google Scholar] [CrossRef]
- Yesiloz, U.; Kirches, E.; Hartmann, C.; Scholz, J.; Kropf, S.; Sahm, F.; Nakamura, M.; Mawrin, C. Frequent akt1e17k mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro. Oncol. 2017, 19, 1088–1096. [Google Scholar] [CrossRef]
- Youngblood, M.W.; Duran, D.; Montejo, J.D.; Li, C.; Omay, S.B.; Ozduman, K.; Sheth, A.H.; Zhao, A.Y.; Tyrtova, E.; Miyagishima, D.F.; et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J. Neurosurg. 2019, 1–10. [Google Scholar] [CrossRef]
- Domingues, P.H.; Teodosio, C.; Otero, A.; Sousa, P.; Ortiz, J.; Macias Mdel, C.; Goncalves, J.M.; Nieto, A.B.; Lopes, M.C.; de Oliveira, C.; et al. Association between inflammatory infiltrates and isolated monosomy 22/del(22q) in meningiomas. PLoS ONE 2013, 8, e74798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, L.; Solito, S.; Masetto, E.; Vettore, M.; Cane, S.; Puppa, A.D.; Mandruzzato, S. Immunosuppressive activity of tumor-infiltrating myeloid cells in patients with meningioma. Oncoimmunology 2018, 7, e1440931. [Google Scholar] [CrossRef] [Green Version]
- Viaene, A.N.; Zhang, B.; Martinez-Lage, M.; Xiang, C.; Tosi, U.; Thawani, J.P.; Gungor, B.; Zhu, Y.; Roccograndi, L.; Zhang, L.; et al. Transcriptome signatures associated with meningioma progression. Acta Neuropathol. Commun. 2019, 7, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadagno, E.; Presta, I.; Maisano, D.; Donato, A.; Pirrone, C.K.; Cardillo, G.; Corrado, S.D.; Mignogna, C.; Mancuso, T.; Donato, G.; et al. Role of macrophages in brain tumor growth and progression. Int. J. Mol. Sci. 2018, 19, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, C.; Dettling, S.; Liu, F.; Ull, A.T.; Warta, R.; Jungk, C.; Roesch, S.; Mock, A.; Sahm, F.; Schmidt, M.; et al. Cytotoxic T cells and their activation status are independent prognostic markers in meningiomas. Clin. Cancer Res. 2019, 25, 5260–5270. [Google Scholar] [CrossRef]
- Domingues, P.H.; Teodosio, C.; Ortiz, J.; Sousa, P.; Otero, A.; Maillo, A.; Barcena, P.; Garcia-Macias, M.C.; Lopes, M.C.; de Oliveira, C.; et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am. J. Pathol. 2012, 181, 1749–1761. [Google Scholar] [CrossRef]
- Proctor, D.T.; Huang, J.; Lama, S.; Albakr, A.; Van Marle, G.; Sutherland, G.R. Tumor-associated macrophage infiltration in meningioma. Neuro-Oncol. Adv. 2019, 1. [Google Scholar] [CrossRef]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. Il-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhang, W.; Yang, P.; Wang, Y.; Li, M.; Zhang, C.; Wang, Z.; Hu, H.; Liu, Y.; Li, Q.; et al. Identification of a 6-cytokine prognostic signature in patients with primary glioblastoma harboring M2 microglia/macrophage phenotype relevance. PLoS One 2015, 10, e0126022. [Google Scholar] [CrossRef]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Galan, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol. 2015, 6, 263. [Google Scholar] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.; Ferluga, S.; Sharma, V.; Futschik, M.; Hilton, D.A.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Proteomic analysis discovers the differential expression of novel proteins and phosphoproteins in meningioma including NEK9, HK2 and SET and deregulation of RNA metabolism. EBioMedicine 2019, 40, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Bassiri, K.; Ferluga, S.; Sharma, V.; Syed, N.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Global proteome and phospho-proteome analysis of merlin-deficient meningioma and schwannoma identifies PDLIM2 as a novel therapeutic target. EBioMedicine 2017, 16, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.J.; Wan, Y.W.; Al-Ouran, R.; Revelli, J.P.; Cardenas, M.F.; Oneissi, M.; Xi, L.; Jalali, A.; Magnotti, J.F.; Muzny, D.M.; et al. Molecular profiling predicts meningioma recurrence and reveals loss of dream complex repression in aggressive tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 21715–21726. [Google Scholar] [CrossRef] [Green Version]
- Kircher, D.A.; Trombetti, K.A.; Silvis, M.R.; Parkman, G.L.; Fischer, G.M.; Angel, S.N.; Stehn, C.M.; Strain, S.C.; Grossmann, A.H.; Duffy, K.L.; et al. AKT1(E17k) activates focal adhesion kinase and promotes melanoma brain metastasis. Mol. Cancer Res. 2019, 17, 1787–1800. [Google Scholar] [CrossRef]
- Kanno, H.; Nishihara, H.; Wang, L.; Yuzawa, S.; Kobayashi, H.; Tsuda, M.; Kimura, T.; Tanino, M.; Terasaka, S.; Tanaka, S. Expression of CD163 prevents apoptosis through the production of granulocyte colony-stimulating factor in meningioma. Neuro. Oncol. 2013, 15, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Domingues, P.; Gonzalez-Tablas, M.; Otero, A.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Goncalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, R.R.; Khairy, R.A. CD44 expression in meningioma and its correlation with proliferation indices. J. Clin. Diagn. Res. 2017, 11, EC12–EC15. [Google Scholar] [CrossRef] [PubMed]
- Kamamoto, D.; Saga, I.; Ohara, K.; Yoshida, K.; Sasaki, H. Association between CD133, CD44, and nestin expression and prognostic factors in high-grade meningioma. World Neurosurg. 2018, 124, E188–E196. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, C.; Maimela, N.R.; Yang, L.; Zhang, Z.; Ping, Y.; Huang, L.; Zhang, Y. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology 2019, 8, 1601478. [Google Scholar] [CrossRef]
- Pelekanou, V.; Villarroel-Espindola, F.; Schalper, K.A.; Pusztai, L.; Rimm, D.L. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in er-positive and -negative cancers. Breast Cancer Res. 2018, 20, 154. [Google Scholar] [CrossRef]
- Kumar, A.T.; Knops, A.; Swendseid, B.; Martinez-Outschoom, U.; Harshyne, L.; Philp, N.; Rodeck, U.; Luginbuhl, A.; Cognetti, D.; Johnson, J.; et al. Prognostic significance of tumor-associated macrophage content in head and neck squamous cell carcinoma: A meta-analysis. Front. Oncol. 2019, 9, 656. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTk (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [Green Version]
- Sevenich, L. Turning “Cold” into “Hot” tumors-opportunities and challenges for radio-immunotherapy against primary and metastatic brain cancers. Front. Oncol. 2019, 9, 163. [Google Scholar] [CrossRef]
- Du, Z.; Abedalthagafi, M.; Aizer, A.A.; McHenry, A.R.; Sun, H.H.; Bray, M.A.; Viramontes, O.; Machaidze, R.; Brastianos, P.K.; Reardon, D.A.; et al. Increased expression of the immune modulatory molecule PD-L1 (CD274) in anaplastic meningioma. Oncotarget 2015, 6, 4704–4716. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.D.; Veliceasa, D.; Lamano, J.B.; Lamano, J.B.; Kaur, G.; Biyashev, D.; Horbinski, C.M.; Kruser, T.J.; Bloch, O. Systemic and local immunosuppression in patients with high-grade meningiomas. Cancer Immunol. Immunother. 2019, 68, 999–1009. [Google Scholar] [CrossRef]
- Proctor, D.T.; Patel, Z.; Lama, S.; Resch, L.; van Marle, G.; Sutherland, G.R. Identification of PD-l2, B7-H3 and CTLA-4 immune checkpoint proteins in genetic subtypes of meningioma. Oncoimmunology 2019, 8, e1512943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.D. PD-L1 expression in meningiomas. J. Clin. Neurosci. 2018, 57, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Erkan, E.P.; Strobel, T.; Dorfer, C.; Sonntagbauer, M.; Weinhausel, A.; Saydam, N.; Saydam, O. Circulating tumor biomarkers in meningiomas reveal a signature of equilibrium between tumor growth and immune modulation. Front. Oncol. 2019, 9, 1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol. Cancer Ther. 2014, 13, 2769–2783. [Google Scholar] [CrossRef] [Green Version]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Holme, J.; Anthony, J. SNP genotyping: The KASP assay. Methods Mol. Biol. 2014, 1145, 75–86. [Google Scholar]
- James, M.F.; Lelke, J.M.; Maccollin, M.; Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Ramesh, V.; Gusella, J.F. Modeling NF2 with human arachnoidal and meningioma cell culture systems: NF2 silencing reflects the benign character of tumor growth. Neurobiol. Dis. 2008, 29, 278–292. [Google Scholar] [CrossRef] [Green Version]
- Hilton, D.A.; Shivane, A.; Kirk, L.; Bassiri, K.; Enki, D.G.; Hanemann, C.O. Activation of multiple growth factor signalling pathways is frequent in meningiomas. Neuropathology 2016, 36, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.V.; Zhuang, H.; Singer, D.; Illsley, C.S.; Kok, W.L.; Sivaraj, K.K.; Gao, Y.; Bolton, C.; Liu, Y.; Zhao, M.; et al. Transit amplifying cells coordinate mouse incisor mesenchymal stem cell activation. Nat. Commun. 2019, 10, 3596. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutated/Total | Detected Frequency % | Frequency in Clark et al., 2016 b % |
|---|---|---|---|
| NF2 (protein loss) a | 49/113 | 43.4 | 33.4 |
| AKT1 E17K | 10/159 | 6.3 | 10.6 |
| KLF4 K409Q | 12/158 | 7.6 | 7.9 |
| PIK3CA H1047R | 2/94 | 2.1 | 1.5 |
| POLR2A Q403K | 3/168 | 1.8 | 2.5 |
| SMARCB1 R377H | 1/139 | 0.7 | Not reported |
| SMO L412F | 3/150 | 2.0 | 3.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, C.L.; Ercolano, E.; Ferluga, S.; Sofela, A.; Dave, F.; Negroni, C.; Kurian, K.M.; Hilton, D.A.; Hanemann, C.O. A Rapid Robust Method for Subgrouping Non-NF2 Meningiomas According to Genotype and Detection of Lower Levels of M2 Macrophages in AKT1 E17K Mutated Tumours. Int. J. Mol. Sci. 2020, 21, 1273. https://doi.org/10.3390/ijms21041273
Adams CL, Ercolano E, Ferluga S, Sofela A, Dave F, Negroni C, Kurian KM, Hilton DA, Hanemann CO. A Rapid Robust Method for Subgrouping Non-NF2 Meningiomas According to Genotype and Detection of Lower Levels of M2 Macrophages in AKT1 E17K Mutated Tumours. International Journal of Molecular Sciences. 2020; 21(4):1273. https://doi.org/10.3390/ijms21041273
Chicago/Turabian StyleAdams, Claire L., Emanuela Ercolano, Sara Ferluga, Agbolahan Sofela, Foram Dave, Caterina Negroni, Kathreena M. Kurian, David A. Hilton, and C. Oliver Hanemann. 2020. "A Rapid Robust Method for Subgrouping Non-NF2 Meningiomas According to Genotype and Detection of Lower Levels of M2 Macrophages in AKT1 E17K Mutated Tumours" International Journal of Molecular Sciences 21, no. 4: 1273. https://doi.org/10.3390/ijms21041273
APA StyleAdams, C. L., Ercolano, E., Ferluga, S., Sofela, A., Dave, F., Negroni, C., Kurian, K. M., Hilton, D. A., & Hanemann, C. O. (2020). A Rapid Robust Method for Subgrouping Non-NF2 Meningiomas According to Genotype and Detection of Lower Levels of M2 Macrophages in AKT1 E17K Mutated Tumours. International Journal of Molecular Sciences, 21(4), 1273. https://doi.org/10.3390/ijms21041273