Emerging Roles of Interleukin-33-responsive Kidney Group 2 Innate Lymphoid Cells in Acute Kidney Injury


Abstract
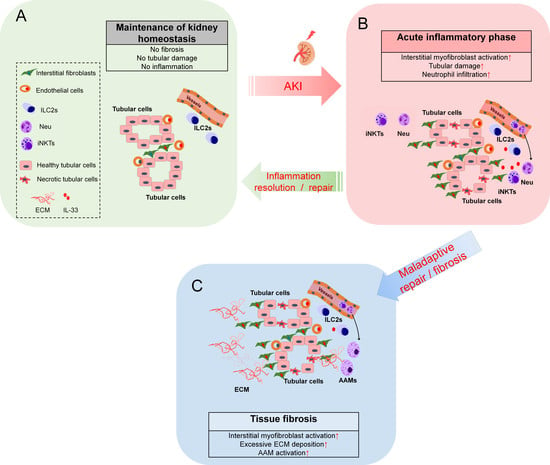
:
1. Introduction
2. Roles of IL-33-Mediated ILC2 Activation in the Kidney
2.1. Characterization of IL-33-Responsive Kidney ILC2s
2.2. Recent Findings on Exogenous IL-33-Mediated ILC2s Activation in Kidney Injury Models
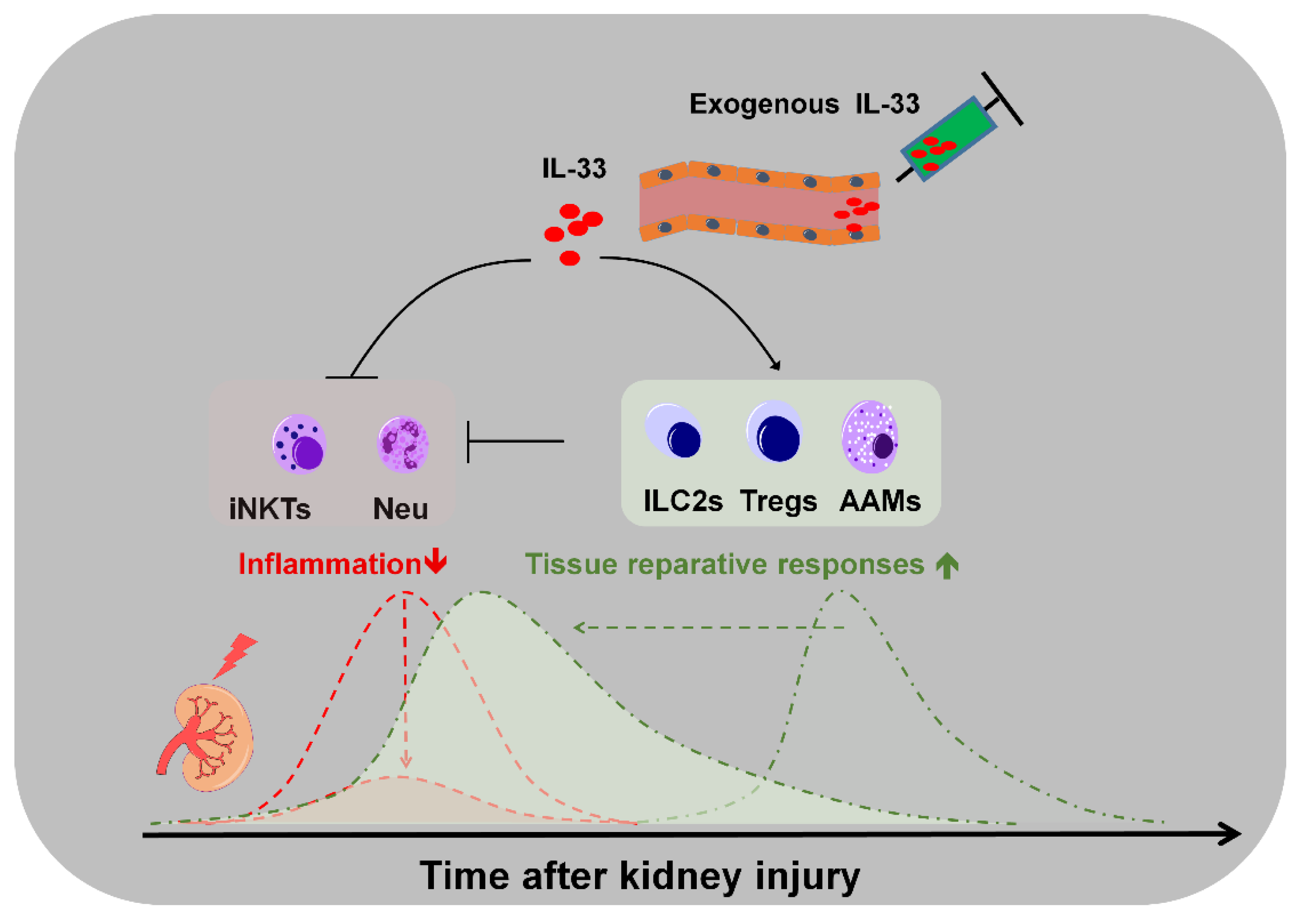
3. Roles of Endogenous IL-33 Versus Exogenous IL-33 in Kidney Injury, Repair, and Fibrosis
4. Potential Applications and Caveats of Therapeutic Targeting IL-33/ST2 Signaling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAMs | alternatively activated macrophages |
| AKI | acute kidney injury |
| AREG | amphiregulin |
| BUN | blood urea nitrogen |
| CKD | chronic kidney disease |
| DM | diabetes mellitus |
| ERK | extracellular signal-regulated kinase |
| ECM | extracellular matrix |
| IGFBP-7 | insulin-like growth factor-binding protein 7 |
| ILC2 | group 2 innate lymphoid cells |
| IL-33 | Interleukin-33 |
| IL-1RacP | IL-1 receptor accessory protein |
| HUVECs | human umbilical vein endothelial cells |
| iNKT | invariant natural killer T-cell |
| IL1RL1 | IL-1 receptor like 1 |
| IRAK-1 | IL-1 receptor-associated kinase-1 |
| IRI | ischemia-reperfusion injury |
| JNK | c-Jun N-terminal kinases |
| KIM-1 | kidney injury molecule-1 |
| MDSCs | myeloid-derived suppressive cells |
| MyD88 | myeloid differentiation primary response 88 |
| NF-κB | nuclear factor kappa B |
| NGAL | neutrophil gelatinase-associated lipocalin |
| PIN1 | peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 |
| SIGIRR | single Ig IL-1-related receptor |
| SCr | serum creatinine |
| TLR | toll-like receptors |
| TIR | toll-interleukin receptor |
| TIMP-2 | tissue inhibitor of metalloproteinases 2 |
| Tregs | regulatory T-cells |
| UUO | unilateral urinary obstruction |
References
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Y.; Pan, Y.; Li, H.; Zheng, S.G.; Su, W. The role of the IL-33/ST2 axis in autoimmune disorders: Friend or foe? Cytokine Growth Factor Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.P. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Talabot-Ayer, D.; Lamacchia, C.; Gabay, C.; Palmer, G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J. Biol. Chem. 2009, 284, 19420–19426. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef] [Green Version]
- Milovanovic, M.; Volarevic, V.; Radosavljevic, G.; Jovanovic, I.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. IL-33/ST2 axis in inflammation and immunopathology. Immunol. Res. 2012, 52, 89–99. [Google Scholar] [CrossRef]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi-Tago, M.; Tago, K.; Hayakawa, M.; Tominaga, S.; Ohshio, T.; Sonoda, Y.; Kasahara, T. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell. Signal. 2008, 20, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechama, M.; Kwon, J.; Wei, S.; Kyi, A.T.; Welner, R.S.; Ben-Dov, I.Z.; Arredouani, M.S.; Asara, J.M.; Chen, C.H.; Tsai, C.Y.; et al. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat. Commun. 2018, 9, 1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulek, K.; Swaidani, S.; Qin, J.; Lu, Y.; Gulen, M.F.; Herjan, T.; Min, B.; Kastelein, R.A.; Aronica, M.; Kosz-Vnenchak, M.; et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J. Immunol. 2009, 182, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Cameron, G.J.M.; Jiang, S.H.; Loering, S.; Deshpande, A.V.; Hansbro, P.M.; Starkey, M.R. Emerging therapeutic potential of group 2 innate lymphoid cells in acute kidney injury. J. Pathol. 2019, 248, 9–15. [Google Scholar] [CrossRef]
- Chen, W.Y.; Li, L.C.; Yang, J.L. Emerging Roles of IL-33/ST2 Axis in Renal Diseases. Int. J. Mol. Sci. 2017, 18, 783. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chang, Y.J.; Su, C.H.; Tsai, T.H.; Chen, S.D.; Hsing, C.H.; Yang, J.L. Upregulation of Interleukin-33 in obstructive renal injury. Biochem. Biophys. Res. Commun. 2016, 473, 1026–1032. [Google Scholar] [CrossRef]
- Akcay, A.; Nguyen, Q.; He, Z.; Turkmen, K.; Won Lee, D.; Hernando, A.A.; Altmann, C.; Toker, A.; Pacic, A.; Ljubanovic, D.G.; et al. IL-33 exacerbates acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Nino, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.; Giraud, S.; Robin, A.; Barra, A.; Bridoux, F.; Ameteau, V.; Hauet, T.; Girard, J.P.; Touchard, G.; Gombert, J.M.; et al. The alarmin concept applied to human renal transplantation: Evidence for a differential implication of HMGB1 and IL-33. PLoS ONE 2014, 9, e88742. [Google Scholar] [CrossRef] [PubMed]
- Rewa, O.; Bagshaw, S.M. Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 2014, 10, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Kellum, J.A.; Lameire, N. The definition of acute kidney injury. Lancet 2018, 391, 202–203. [Google Scholar] [CrossRef] [Green Version]
- Dennen, P.; Parikh, C.R. Biomarkers of acute kidney injury: Can we replace serum creatinine? Clin. Nephrol. 2007, 68, 269–278. [Google Scholar] [CrossRef]
- Aregger, F.; Uehlinger, D.E.; Witowski, J.; Brunisholz, R.A.; Hunziker, P.; Frey, F.J.; Jorres, A. Identification of IGFBP-7 by urinary proteomics as a novel prognostic marker in early acute kidney injury. Kidney Int. 2014, 85, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Huen, S.C.; Parikh, C.R. Molecular phenotyping of clinical AKI with novel urinary biomarkers. Am. J. Physiol. Renal Physiol. 2015, 309, F406–F413. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.R.; Parikh, C.R. Biomarkers of Acute and Chronic Kidney Disease. Annu. Rev. Physiol. 2019, 81, 309–333. [Google Scholar] [CrossRef]
- Takahashi, S.; Nakasatomi, M.; Takei, Y.; Ikeuchi, H.; Sakairi, T.; Kaneko, Y.; Hiromura, K.; Nojima, Y.; Maeshima, A. Identification of Urinary Activin A as a Novel Biomarker Reflecting the Severity of Acute Kidney Injury. Sci. Rep. 2018, 8, 5176. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, A.; Faubel, S.; Askenazi, D.J.; Cerda, J.; Fissell, W.H.; Heung, M.; Humphreys, B.D.; Koyner, J.L.; Liu, K.D.; Mour, G.; et al. Clinical Use of the Urine Biomarker [TIMP-2] x [IGFBP7] for Acute Kidney Injury Risk Assessment. Am. J. Kidney Dis. 2016, 68, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Moore, P.K.; Hsu, R.K.; Liu, K.D. Management of Acute Kidney Injury: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 72, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Acute Dialysis Quality Initiative Consensus, X.W.G. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Fu, H.; Liu, S.; Zhang, L.; Xiao, L.; Bastacky, S.I.; Liu, Y. Early activation of fibroblasts is required for kidney repair and regeneration after injury. FASEB J. 2019, 33, 12576–12587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Harris, D.C.; Wang, Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015, 30, 183–194. [Google Scholar] [CrossRef]
- Kellum, J.A.; Sileanu, F.E.; Bihorac, A.; Hoste, E.A.; Chawla, L.S. Recovery after Acute Kidney Injury. Am. J. Respir. Crit. Care Med. 2017, 195, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Martin-Cleary, C.; Garcia-Bermejo, L.; Moreno, J.A.; Ruiz-Ortega, M.; Draibe, J.; Cruzado, J.M.; Garcia-Gonzalez, M.A.; et al. Translational value of animal models of kidney failure. Eur. J. Pharmacol. 2015, 759, 205–220. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, Z. Mouse model of ischemic acute kidney injury: Technical notes and tricks. Am. J. Physiol. Renal Physiol. 2012, 303, F1487–F1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Klimova, E.; Aparicio-Trejo, O.E.; Tapia, E.; Pedraza-Chaverri, J. Unilateral Ureteral Obstruction as a Model to Investigate Fibrosis-Attenuating Treatments. Biomolecules 2019, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Braun, H.; Afonina, I.S.; Mueller, C.; Beyaert, R. Dichotomous function of IL-33 in health and disease: From biology to clinical implications. Biochem. Pharmacol. 2018, 148, 238–252. [Google Scholar] [CrossRef]
- Afferni, C.; Buccione, C.; Andreone, S.; Galdiero, M.R.; Varricchi, G.; Marone, G.; Mattei, F.; Schiavoni, G. The Pleiotropic Immunomodulatory Functions of IL-33 and Its Implications in Tumor Immunity. Front. Immunol. 2018, 9, 2601. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. ‘Repair’ Treg Cells in Tissue Injury. Cell Physiol. Biochem. 2017, 43, 2155–2169. [Google Scholar] [CrossRef] [Green Version]
- Riedel, J.H.; Becker, M.; Kopp, K.; Duster, M.; Brix, S.R.; Meyer-Schwesinger, C.; Kluth, L.A.; Gnirck, A.C.; Attar, M.; Krohn, S.; et al. IL-33-Mediated Expansion of Type 2 Innate Lymphoid Cells Protects from Progressive Glomerulosclerosis. J. Am. Soc. Nephrol. 2017, 28, 2068–2080. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, Y.; Niu, Z.; Wang, C.; Wang, R.; Zhang, Z.; Chen, T.; Wang, X.M.; Li, Q.; Lee, V.W.S.; et al. Potentiating Tissue-Resident Type 2 Innate Lymphoid Cells by IL-33 to Prevent Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2018, 29, 961–976. [Google Scholar] [CrossRef]
- Sabapathy, V.; Cheru, N.T.; Corey, R.; Mohammad, S.; Sharma, R. A Novel Hybrid Cytokine IL233 Mediates regeneration following Doxorubicin-Induced Nephrotoxic Injury. Sci. Rep. 2019, 9, 3215. [Google Scholar] [CrossRef] [Green Version]
- Stremska, M.E.; Jose, S.; Sabapathy, V.; Huang, L.; Bajwa, A.; Kinsey, G.R.; Sharma, P.R.; Mohammad, S.; Rosin, D.L.; Okusa, M.D.; et al. IL233, A Novel IL-2 and IL-33 Hybrid Cytokine, Ameliorates Renal Injury. J. Am. Soc. Nephrol. 2017, 28, 2681–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Xu, F.; Wen, X.J.; Liu, H.Z.; Wang, H.B.; Zhong, J.Y.; Yang, C.X.; Zhang, B. Interleukin-33 signaling contributes to renal fibrosis following ischemia reperfusion. Eur. J. Pharmacol. 2017, 812, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mao, L.; Wu, X.; Wu, T.; Liu, W.; Yang, Y.; Zhang, T.; Xu, Y. BRG1 regulates endothelial-derived IL-33 to promote ischemia-reperfusion induced renal injury and fibrosis in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2551–2561. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Yu, T.; Yan, B.; Li, H. Interleukin33 promotes obstructive renal injury via macrophages. Mol. Med. Rep. 2019, 20, 1353–1362. [Google Scholar] [CrossRef]
- Park, G.H.; Shinn, H.K.; Kang, J.H.; Na, W.J.; Kim, Y.H.; Park, C.S. Anti-interleukin-33 Reduces Ovalbumin-Induced Nephrotoxicity and Expression of Kidney Injury Molecule-1. Int. Neurourol. J. 2016, 20, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Tait Wojno, E.D.; Beamer, C.A. Isolation and Identification of Innate Lymphoid Cells (ILCs) for Immunotoxicity Testing. Methods Mol. Biol. 2018, 1803, 353–370. [Google Scholar] [CrossRef]
- Chen, W.Y.; Yang, J.L.; Wu, Y.H.; Li, L.C.; Li, R.F.; Chang, Y.T.; Dai, L.H.; Wang, W.C.; Chang, Y.J. IL-33/ST2 axis mediates hyperplasia of intrarenal urothelium in obstructive renal injury. Exp. Mol. Med. 2018, 50, 36. [Google Scholar] [CrossRef] [Green Version]
- Cameron, G.J.M.; Cautivo, K.M.; Loering, S.; Jiang, S.H.; Deshpande, A.V.; Foster, P.S.; McKenzie, A.N.J.; Molofsky, A.B.; Hansbro, P.M.; Starkey, M.R. Group 2 Innate Lymphoid Cells Are Redundant in Experimental Renal Ischemia-Reperfusion Injury. Front. Immunol. 2019, 10, 826. [Google Scholar] [CrossRef]
- Vely, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef] [Green Version]
- Stier, M.T.; Zhang, J.; Goleniewska, K.; Cephus, J.Y.; Rusznak, M.; Wu, L.; Van Kaer, L.; Zhou, B.; Newcomb, D.C.; Peebles, R.S., Jr. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 2018, 215, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Ferhat, M.; Robin, A.; Giraud, S.; Sena, S.; Goujon, J.M.; Touchard, G.; Hauet, T.; Girard, J.P.; Gombert, J.M.; Herbelin, A.; et al. Endogenous IL-33 Contributes to Kidney Ischemia-Reperfusion Injury as an Alarmin. J. Am. Soc. Nephrol. 2018, 29, 1272–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardman, C.; Ogg, G. Interleukin-33, friend and foe in type-2 immune responses. Curr. Opin. Immunol. 2016, 42, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Niu, Z.; Tan, J.; Yang, J.; Liu, Y.; Ma, H.; Lee, V.W.; Sun, S.; Song, X.; Guo, M.; et al. IL-25 Elicits Innate Lymphoid Cells and Multipotent Progenitor Type 2 Cells That Reduce Renal Ischemic/Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 2199–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stremska, M.E.; Dai, C.; Venkatadri, R.; Wang, H.; Sabapathy, V.; Kumar, G.; Jose, S.; Mohammad, S.; Sung, S.J.; Fu, S.M.; et al. IL233, an IL-2-IL-33 hybrid cytokine induces prolonged remission of mouse lupus nephritis by targeting Treg cells as a single therapeutic agent. J. Autoimmun. 2019, 102, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, V.; Stremska, M.E.; Mohammad, S.; Corey, R.L.; Sharma, P.R.; Sharma, R. Novel Immunomodulatory Cytokine Regulates Inflammation, Diabetes, and Obesity to Protect From Diabetic Nephropathy. Front. Pharmacol. 2019, 10, 572. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, D.C.; Melo, P.H.; Pineros, A.R.; Ferreira, R.G.; Colon, D.F.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017, 8, 14919. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel ‘alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R.; et al. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer. Am. J. Physiol. Renal Physiol. 2018, 314, F356–F366. [Google Scholar] [CrossRef]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Tao, L.; Chen, C.; Song, H.; Li, Z.; Gao, Y.; Nie, J.; Piccioni, M.; Shi, G.; Li, B. The Deubiquitinase USP17 Regulates the Stability and Nuclear Function of IL-33. Int. J. Mol. Sci. 2015, 16, 27956–27966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, J.; Rochman, M.; Miracle, C.E.; Habel, J.E.; Brusilovsky, M.; Caldwell, J.M.; Rymer, J.K.; Rothenberg, M.E. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat. Commun. 2018, 9, 3244. [Google Scholar] [CrossRef] [PubMed]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Girard, J.P. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef]
- Sehnine, M.; Ferhat, M.; Sena, S.; Gombert, J.M.; Goujon, J.M.; Thierry, A.; Touchard, G.; Hauet, T.; Herbelin, A.; Hadjadj, S. IL-33 receptor ST2 deficiency attenuates renal ischaemia-reperfusion injury in euglycaemic, but not streptozotocin-induced hyperglycaemic mice. Diabetes Metab. 2018, 44, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.Y.; Hong, J.; Gannon, J.; Kakkar, R.; Lee, R.T. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc. Natl. Acad. Sci. USA 2015, 112, 7249–7254. [Google Scholar] [CrossRef] [Green Version]
- Schulze Blasum, B.; Schroter, R.; Neugebauer, U.; Hofschroer, V.; Pavenstadt, H.; Ciarimboli, G.; Schlatter, E.; Edemir, B. The kidney-specific expression of genes can be modulated by the extracellular osmolality. FASEB J. 2016, 30, 3588–3597. [Google Scholar] [CrossRef] [Green Version]
- Krishack, P.A.; Louviere, T.J.; Decker, T.S.; Kuzel, T.G.; Greenberg, J.A.; Camacho, D.F.; Hrusch, C.L.; Sperling, A.I.; Verhoef, P.A. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Species | ILC Subsets (Markers) | Characteristics | Tissue Localization | Reference |
|---|---|---|---|---|
| Human | Total ILCs (Lin-CD127+CD161+); ILC2s (Lin-CD127+CD161+CRTH2+); ILC3s (NKp44+, and NKp44-CRTH2-CD117+); ILC1s (CRTH2-CD117-NKp44-) | * ILC3 constitute ~55% of total ILCs; * ILC2 constitute ~30% of total ILCs; * ILC1 constitute ~10% of total ILCs | Riedel et al. [48] | |
| Mouse | ILC2s CD45+Lin-CD127+CD90.2+ST2+ | *Kidney ILC2s constitutively express IL-5 | IL-5+ILC2s localize the major vasculature in healthy kidney | Cameron et al. [59] |
| Mouse | ILC2s (CD45+Lin-CD127+GATA3+ | * Mouse ILC2s constitute ~80% of total ILC2s | ILC2s localized in glomerular and tubulointerstitial compartments | Riedel et al. [48] |
| Mouse | ILC2s (CD45+Lin-CD127+GATA3+ST2+) | * IRI alone did not altered ILC2s | Cao et al. [49] | |
| * Exogenous IL-33 expands ILC2s | ||||
| Mouse | ILC2s (CD45+Lin-ST2+) | * Adoptive transfer of ex vivo expanded spleen-derived ILC2s by IL-233 hybrid protects the against IRI | Stremska et al. [51] | |
| Mouse | ILC2s (CD45+Lin-ST2+CD90+) | * IL233 expands spleen and kidney ILC2s | Sabapathy et al. [50] | |
| Mouse | ILC2s (CD45+Lin-CD90.2+ GATA3+) | * Mouse ILC2s constitute ~70% of total ILCs; | Chen et al. [58] | |
| * ILC2s were expended 2 fold following UUO |
| Animals | Model | Key Findings | Reference |
| Endogenous IL-33 | |||
| IL33Gt/Gt | IRI | Capillary CD31+ endothelial cells are the source of IL-33 | Ferhat et al. [62] |
| IL-33 knockout is protective to IRI | |||
| Reduced myeloid cell infiltration in IL-33 knockout mice | |||
| Impaired iNKT recruitment and function in the IL-33 knockout mice | |||
| Early IL-33 release (1–6 h post injury) is not necessary for myeloid recruitment after IRI | |||
| IL-33-mediated iNKT activation contributes to neutrophil recruitment during the amplification phase of kidney injury | |||
| Exogenous IL-33 treatment | |||
| mouse IL-33 (0.3 μg/mice for 5 days); C57BL/6 | IRI | Exogenous IL-33 protects mice against IRI (pre-treatment or post-treatment) | Cao et al. [49] |
| Exogenous IL-33 decreased GR-1+ myeloid cells post-IRI | |||
| Exogenous IL-33 induced Th2 cytokines, ILC2s, Tregs, and AAMs in vivo | |||
| IL-33 boosts kidney-resident ILC2 proliferation in vivo | |||
| Treg did not contribute to IL-33-mediated renoprotection in IRI mice | |||
| GW2580-mediated AAM depletion abolished IL-33-mediated renoprotection in IRI mice | |||
| Anti-CD90-mediated ILC depletion abolished IL-33-mediated renoprotection in IRI mice | |||
| Adoptive transfer of ILC2 protects mice from IRI | |||
| AREG mediates renoprotective function of ILC2s | |||
| IL-33 and ILC2s are protective post-IRI | |||
| mouse IL233 (66pmol /mice for 5 days); C57BL/6J and Balb/cJ | IRI Doxorubicin Cisplatin | Exogenous IL-2 combined with IL-33 (IL233) is protective post-IRI | Stremska et al. [51]; Sabapathy et al. [50] |
| IL233 promotes the expansion of ILC2s and Tregs, which is required for the renal protective functions | |||
| IL233 is renal protective in IRI-, cisplatin-, and doxorubicin-induced nephrotoxicity | |||
| mouse IL-33 | IRI | Exogenous IL-33 promotes IRI-induced fibrosis | Liang et al. [53] |
| sST2 attenuates IRI-induced renal injury and fibrosis | |||
| mouse IL-33 (0.4μg/mice for 4 days); C57BL/6 and Balb/c | Adriamycin (Doxorubicin) | ST2+ILC2s are the major ILC2 population in human and mouse kidneys | Riedel et al. [48] |
| Exogenous IL-33 induces sustained ILC2 expansion in the kidneys | |||
| Exogenous IL-33 ameliorates Adrimycin-induced kidney injury | |||
| Eosinophils are required for IL-33-mediated tissue protection | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-Y.; Li, L.-C.; Wu, Y.-H.; Yang, J.-L.; Tzeng, H.-T. Emerging Roles of Interleukin-33-responsive Kidney Group 2 Innate Lymphoid Cells in Acute Kidney Injury. Int. J. Mol. Sci. 2020, 21, 1544. https://doi.org/10.3390/ijms21041544
Chen W-Y, Li L-C, Wu Y-H, Yang J-L, Tzeng H-T. Emerging Roles of Interleukin-33-responsive Kidney Group 2 Innate Lymphoid Cells in Acute Kidney Injury. International Journal of Molecular Sciences. 2020; 21(4):1544. https://doi.org/10.3390/ijms21041544
Chicago/Turabian StyleChen, Wei-Yu, Lung-Chih Li, Yi-Hsiu Wu, Jenq-Lin Yang, and Hong-Tai Tzeng. 2020. "Emerging Roles of Interleukin-33-responsive Kidney Group 2 Innate Lymphoid Cells in Acute Kidney Injury" International Journal of Molecular Sciences 21, no. 4: 1544. https://doi.org/10.3390/ijms21041544
APA StyleChen, W. -Y., Li, L. -C., Wu, Y. -H., Yang, J. -L., & Tzeng, H. -T. (2020). Emerging Roles of Interleukin-33-responsive Kidney Group 2 Innate Lymphoid Cells in Acute Kidney Injury. International Journal of Molecular Sciences, 21(4), 1544. https://doi.org/10.3390/ijms21041544