Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs


Abstract
:1. Introduction
2. Results
2.1. Transcripts Assembly and Identification of LincRNAs
2.2. Characterization of Protein-Coding Transcripts and Identified LincRNAs
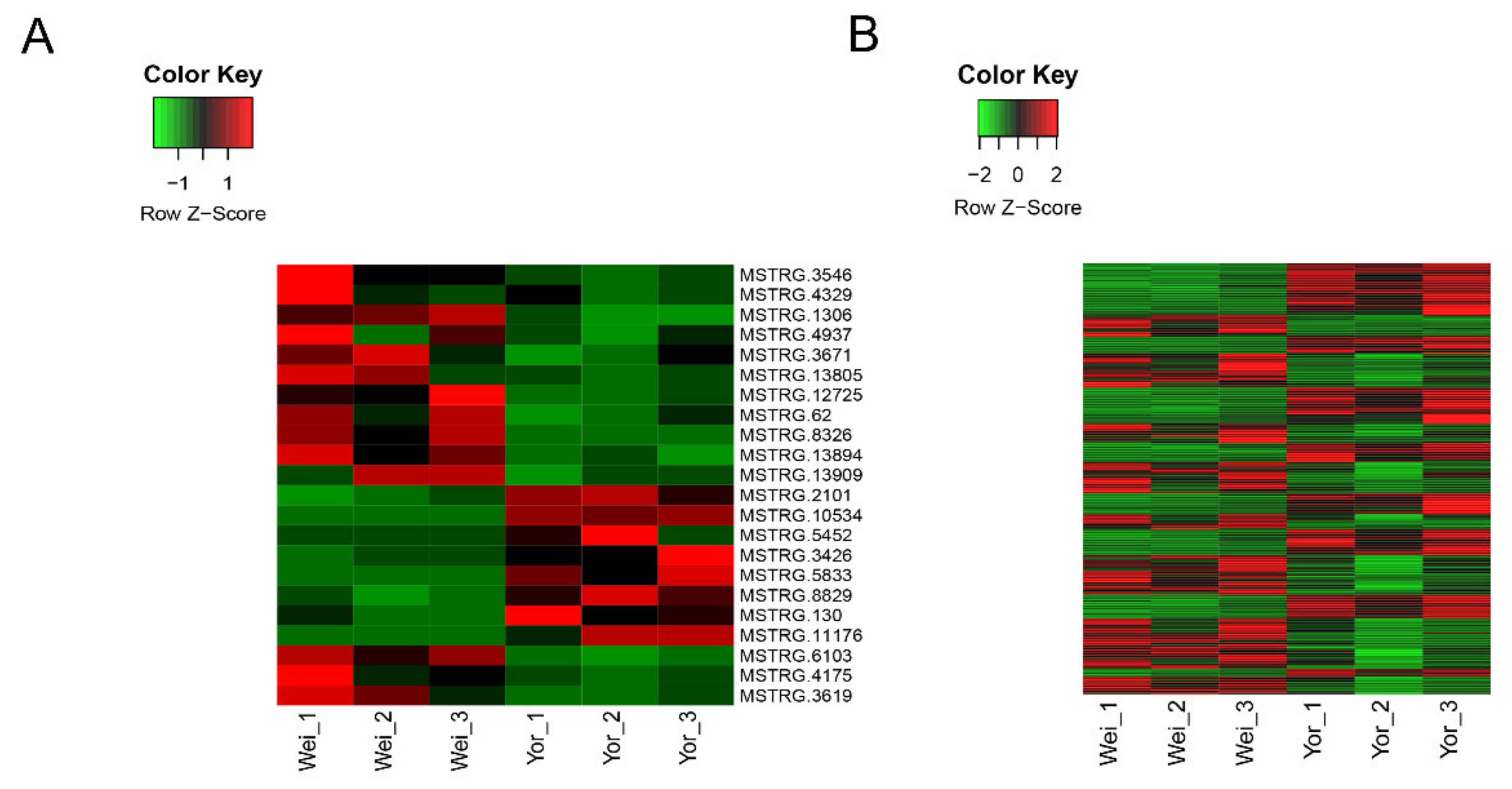
2.3. Differential Expression Analysis of LincRNAs and Protein-Coding Genes
2.4. Association Analysis Between QTL Sites and DE LincRNAs Location
2.5. Prediction of Target Genes of DE LincRNAs
2.6. Functional Enrichment Analysis of the PTGs of DE LincRNAs
2.7. Expression Regulation Analysis of DE LincRNAs and their DEPTGs
2.8. Correlation Validation of LincRNAs and their PTGs by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Data Acquisition
4.2. RNA-Seq Reads Mapping and Transcriptome Assembly
- (1)
- fastqc -o outdir -t threads fastq1 fastq2.
- (2)
- hisat2 -p 8 --dta --known-splicesite-infile splicesites.txt –x genome -1 sample_1_1_clean.fa -2 sample_1_2_clean.fa –S sample_1.sam
- (3)
- stringtie --merge -p 8 -G genome_reference.gtf -o stringtie_merged.gtf stringtie_merge.txt.
4.3. Pipeline for LincRNA Identification
4.4. Comparisons Between LincRNAs and Protein-Coding Transcripts
4.5. Analysis of Differentially Expressed LincRNAs and Protein Coding Genes
4.6. QTLs Analysis of DE LincRNAs
4.7. Prediction of PTGs of LincRNAs
4.8. Gene Ontology and Pathway Analysis
4.9. Correlation Validation Between LincRNAs and PTGs by Real-Time Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Z.Y.; Xu, H.T.; Li, Y.Y.; Wen, L.; Li, J.Q.; Lu, G.H.; Li, X.Y. Climate and drought risk regionalisation in China based on probabilistic aridity and drought index. Sci. Total Envrion. 2018, 612, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Fortin, A.; Robertson, W.M.; Tong, A.K.W. The eating quality of Canadian pork and its relationship with intramuscular fat. Meat Sci. 2005, 69, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Pietruszka, A.; Jacyno, E.; Kawecka, M.; Biel, W. The Relation between Intramuscular Fat Level in the Longissimus Muscle and the Quality of Pig Carcasses and Meat. Ann. Anim. Sci. 2015, 15, 1031–1041. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, X.; Sairam, M.R. Novel Genes of Visceral Adiposity: Identification of Mouse and Human Mesenteric Estrogen-Dependent Adipose (MEDA)-4 Gene and Its Adipogenic Function. Endocrinology 2012, 153, 2665–2676. [Google Scholar] [CrossRef] [Green Version]
- Kaaman, M.; Rydén, M.; Axelsson, T.; Nordström, E.; Sicard, A.; Bouloumié, A.; Langin, D.; Arner, P.; Dahlman, I. ALOX5AP expression, but not gene haplotypes, is associated with obesity and insulin resistance. Int. J. Obes. 2006, 30, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Sun, W.; Han, H.; Chu, W.; Zhang, L.; Chen, J. miR-130a regulates differential lipid accumulation between intramuscular and subcutaneous adipose tissues of pigs via suppressing PPARG expression. Gene 2017, 636, 23–29. [Google Scholar] [CrossRef]
- Li, C.; Zheng, H.; Hou, W.; Bao, H.; Xiong, J.; Che, W.; Gu, Y.; Sun, H.; Liang, P. Long non-coding RNA linc00645 promotes TGF-beta-induced epithelial-mesenchymal transition by regulating miR-205-3p-ZEB1 axis in glioma. Cell Death Dis. 2019, 10, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.-W.; Chen, Y.-Q. Potential Pathological and Functional Links Between Long Noncoding RNAs and Hematopoiesis. Sci. Signal. 2013, 6, re5. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-q.; Wang, Y.; Xiong, Y.; Chen, X.-C.; Ma, M.-l.; Cai, R.; Gao, Y.; Sun, Y.-m.; Yang, G.-S.; Pang, W.-J. Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Sci. Rep. 2016, 6, 21865. [Google Scholar] [CrossRef]
- De Lima, D.S.; Cardozo, L.E.; Maracaja-Coutinho, V.; Suhrbier, A.; Mane, K.; Jeffries, D.; Silveira, E.L.V.; Amaral, P.P.; Rappuoli, R.; de Silva, T.I.; et al. Long noncoding RNAs are involved in multiple immunological pathways in response to vaccination. Proc. Natl. Acad. Sci. USA 2019, 116, 17121–17126. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Chen, L.; Chen, G.; Zou, C.; Li, J.; Li, M.; Fang, C.; Li, C. Identification and Functional Prediction of Long Intergenic Non-coding RNAs Related to Subcutaneous Adipose Development in Pigs. Front. Genet. 2019, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cheng, X.; Shi, G.; Zou, C.; Chen, L.; Li, J.; Li, M.; Fang, C.; Li, C. Transcriptome Analysis Reveals the Effect of Long Intergenic Noncoding RNAs on Pig Muscle Growth and Fat Deposition. Biomed. Res. Int. 2019, 2019, 2951427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Sun, Y.; Cai, R.; Wang, G.; Shu, X.; Pang, W. Long noncoding RNA: Multiple players in gene expression. BMB Rep. 2018, 51, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, C.; Jin, E.; Gu, Y.; Li, S.; Li, Q. Identification of differentially expressed genes in longissimus dorsi muscle between Wei and Yorkshire pigs using RNA sequencing. Genes Genom. 2018, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Qian, K.; Wang, C. Discovery of porcine miRNA-196a/b may influence porcine adipogenesis in longissimus dorsi muscle by miRNA sequencing. Anim. Genet. 2017, 48, 175–181. [Google Scholar] [CrossRef]
- Zou, C.; Li, L.; Cheng, X.; Li, C.; Fu, Y.; Fang, C.; Li, C. Identification and Functional Analysis of Long Intergenic Non-coding RNAs Underlying Intramuscular Fat Content in Pigs. Front. Genet. 2018, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Wu, Y.; Yang, Y.; Yang, Y.T.; Wang, Z.; Yuan, J.; Yang, Y.; Hua, C.; Fan, X.; Niu, G.; et al. Comprehensive analysis of long non-coding RNAs highlights their spatio-temporal expression patterns and evolutional conservation in Sus scrofa. Sci. Rep. 2017, 7, 43166. [Google Scholar] [CrossRef]
- Miao-Chih, T.; Ohad, M.; Yue, W.; Nima, M.; Wang, J.K.; Fei, L.; Yang, S.; Eran, S.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Massolini, G.; Calleri, E. Survey of binding properties of fatty acid-binding proteins. Chromatographic methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 797, 255–268. [Google Scholar] [CrossRef]
- Mercade, A.; Estelle, J.; Perez-Enciso, M.; Varona, L.; Silio, L.; Noguera, J.L.; Sanchez, A.; Folch, J.M. Characterization of the porcine acyl-CoA synthetase long-chain 4 gene and its association with growth and meat quality traits. Anim. Genet. 2006, 37, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; Lei, O.L.; Coleman, R.A. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J. Lipid Res. 2006, 47, 2004–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-F.; Yuan, Z.-Q.; Song, D.-G.; Zhou, X.-H.; Wang, Y.-Z. Effects of cannabinoid receptor 1 (brain) on lipid accumulation by transcriptional control of CPT1A and CPT1B. Anim. Genet. 2014, 45, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Sohara, E.; Rai, T.; Ikawa, M.; Okabe, M.; Sasaki, S.; Uchida, S.; Verkman, A.S. Progressive adipocyte hypertrophy in aquaporin-7-deficient mice: Adipocyte glycerol permeability as a novel regulator of fat accumulation. J. Biol. Chem. 2005, 280, 15493–15496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, N.; Funahashi, T.; Hibuse, T.; Nagasawa, A.; Kishida, K.; Kuriyama, H.; Nakamura, T.; Kihara, S.; Shimomura, I.; Matsuzawa, Y. Adaptation to fasting by glycerol transport through aquaporin 7 in adipose tissue. Proc. Natl. Acad. Sci. USA 2004, 101, 17801–17806. [Google Scholar] [CrossRef] [Green Version]
- Ribon, V.; Johnson, J.H.; Camp, H.S.; Saltiel, A.R. Thiazolidinediones and insulin resistance: Peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc. Natl. Acad. Sci. USA 1998, 95, 14751–14756. [Google Scholar] [CrossRef]
- Spurlock, M.E.; Gabler, N.K. The development of porcine models of obesity and the metabolic syndrome. J. Nutr. 2008, 138, 397–402. [Google Scholar] [CrossRef]
- Lunney, J.K. Advances in swine biomedical model genomics. Int. J. Biol. Sci. 2007, 3, 179–184. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Li, S.; Deng, L.; Guan, Y.; Chen, D.; Yuan, X.; Xia, T.; He, X.; Shan, Y.; Li, C. Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig. Genes 2017, 8, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bumgarner, S.L.; Dowell, R.D.; Paula, G.; Gifford, D.K.; Fink, G.R. Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc. Natl. Acad. Sci. USA 2009, 106, 18321–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, S.; Lin, B.; Chou, T.; Arroyo, K.; Sun, S. LncRNA Jpx induces Xist expression in mice using both trans and cis mechanisms. PLoS Genet. 2018, 14, e1007378. [Google Scholar] [CrossRef]
- Hong, S.H.; Kwon, J.T.; Kim, J.; Jeong, J.; Kim, J.; Lee, S.; Cho, C. Profiling of testis-specific long noncoding RNAs in mice. BMC Genom. 2018, 19, 539. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chen, D.; Zhang, T.; Duan, A.; Zhang, J.; He, C. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2018, 25, 465–476. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shin, D.; Song, K.D. Dominance effects of ion transport and ion transport regulator genes on the final weight and backfat thickness of Landrace pigs by dominance deviation analysis. Genes Genom. 2018, 40, 1331–1338. [Google Scholar] [CrossRef]
- Xiao, B.; Zhang, W.; Chen, L.; Hang, J.; Wang, L.; Rong, Z.; Yang, L.; Chen, J.; Qiang, M.; Sun, Z. Analysis of the miRNA–mRNA–lncRNA network in human estrogen receptor-positive and estrogen receptor-negative breast cancer based on TCGA data. Gene 2018, 658, 28–35. [Google Scholar] [CrossRef]
- Liesenfeld, D.B.; Dmitry, G.; Fahrmann, J.F.; Mariam, S.; Dominique, S.; Reka, T.; Nina, H.; Jürgen, B.H.; Petra, S.K.; Biljana, G. Metabolomics and transcriptomics identify pathway differences between visceral and subcutaneous adipose tissue in colorectal cancer patients: The ColoCare study. Am. J. Clin. Nutr. 2016, 102, 433–443. [Google Scholar] [CrossRef]
- Ohto, T.; Uozumi, N.; Hirabayashi, T.; Shimizu, T. Identification of novel cytosolic phospholipase A(2)s, murine cPLA(2){delta}, {epsilon}, and {zeta}, which form a gene cluster with cPLA(2){beta}. J. Biol. Chem. 2005, 280, 24576–24583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Parsons, W.H.; Kamat, S.S.; Cravatt, B.F. A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat. Chem. Biol. 2016, 12, 669–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piomelli, D. A fatty gut feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.A.S.; Tsuboi, K.; Uyama, T.; Ueda, N. New players in the fatty acyl ethanolamide metabolism. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2014, 86, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Kang, J.; Dai, J.; Tang, M.; Wang, Q.; Zhang, H.; Guo, W.; Sun, R.; Yu, H. AGXT2L1 is down-regulated in heptocellular carcinoma and associated with abnormal lipogenesis. J. Clin. Pathol. 2016, 69, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Poppelreuther, M.; Ehehalt, R.; Fullekrug, J. Overexpressed FATP1, ACSVL4/FATP4 and ACSL1 increase the cellular fatty acid uptake of 3T3-L1 adipocytes but are localized on intracellular membranes. PLoS ONE 2012, 7, e45087. [Google Scholar] [CrossRef]
- Li, Q.; Tao, Z.; Shi, L.; Ban, D.; Zhang, B.; Yang, Y.; Zhang, H.; Wu, C. Expression and genome polymorphism of ACSL1 gene in different pig breeds. Mol. Biol. Rep. 2012, 39, 8787–8792. [Google Scholar] [CrossRef]
- Li, X.; Kim, S.W.; Choi, J.S.; Lee, Y.M.; Lee, C.K.; Choi, B.H.; Kim, T.H.; Choi, Y.I.; Kim, J.J.; Kim, K.S. Investigation of porcine FABP3 and LEPR gene polymorphisms and mRNA expression for variation in intramuscular fat content. Mol. Biol. Rep. 2010, 37, 3931–3939. [Google Scholar] [CrossRef]
- Zhang, Y.; Kent, J.W.; Lee, A.; Cerjak, D.; Ali, O.; Diasio, R.; Olivier, M.; Blangero, J.; Carless, M.A.; Kissebah, A.H. Fatty acid binding protein 3 (fabp3) is associated with insulin, lipids and cardiovascular phenotypes of the metabolic syndrome through epigenetic modifications in a Northern European family population. BMC Med. Genom. 2013, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Madeira, A.; Camps, M.; Zorzano, A.; Moura, T.F.; Soveral, G. Biophysical assessment of human aquaporin-7 as a water and glycerol channel in 3T3-L1 adipocytes. PLoS ONE 2013, 8, e83442. [Google Scholar] [CrossRef]
- Lebeck, J.; Sondergaard, E.; Nielsen, S. Increased AQP7 abundance in skeletal muscle from obese men with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E367–E373. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Huang, C.J.; Liu, M.W.; Chang, H.M.; Chen, Y.J.; Tai, T.Y.; Chuang, L.M. Cloning, mapping, and characterization of the human sorbin and SH3 domain containing 1 (SORBS1) gene: A protein associated with c-Abl during insulin signaling in the hepatoma cell line Hep3B. Genomics 2001, 74, 12–20. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, W.J.; Huang, K.C.; Lee, K.C.; Chao, C.L.; Chen, C.L.; Tai, T.Y.; Chuang, L.M. mRNA levels of the insulin-signaling molecule SORBS1 in the adipose depots of nondiabetic women. Obes. Res. 2003, 11, 586–590. [Google Scholar] [CrossRef]
- Xiao, H.; Yuan, Z.; Guo, D.; Hou, B.; Yin, C.; Zhang, W.; Li, F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genom. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Seo, H.D.; Hennighausen, L.; Lee, D.; Kang, K. Octopus-toolkit: A workflow to automate mining of public epigenomic and transcriptomic next-generation sequencing data. Nucleic Acids Res. 2018, 46, e53. [Google Scholar] [CrossRef] [Green Version]
- Keel, B.N.; Snelling, W.M. Comparison of Burrows-Wheeler Transform-Based Mapping Algorithms Used in High-Throughput Whole-Genome Sequencing: Application to Illumina Data for Livestock Genomes. Front. Genet. 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, K.; Yong, Z.; Zhi-Qiang, Y.; Xiao-Qiao, L.; Shu-Qi, Z.; Liping, W.; Ge, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345. [Google Scholar]
- Prakash, A.; Jeffryes, M.; Bateman, A.; Finn, R.D. The HMMER Web Server for Protein Sequence Similarity Search. Curr. Protoc. Bioinform. 2017, 60, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Perkins, E.J.; Deng, Y. Batch Blast Extractor: An automated blastx parser application. BMC Genom. 2008, 9, S10. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxf. Engl.) 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Dan, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhao, X.; Li, Q. Genome-wide identification and characterization of long intergenic noncoding RNAs and their potential association with larval development in the Pacific oyster. Sci. Rep. 2016, 6, 20796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fu, L.; Koganti, P.P.; Wang, L.; Hand, J.M.; Ma, H.; Yao, J. Identification and Functional Prediction of Large Intergenic Noncoding RNAs (lincRNAs) in Rainbow Trout (Oncorhynchus mykiss). Mar. Biotechnol. 2016, 18, 271–282. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics (Oxf. Engl.) 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kersey, P.J.; Allen, J.E.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; Hughes, D.S.; Humphrey, J.; Kerhornou, A.; Khobova, J. Ensembl Genomes 2013: Scaling up access to genome-wide data. Nucleic Acids Res. 2014, 42, 546–552. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Accession Number | Raw Reads | Clean Reads | Mapped Reads | Mapping Ratio | Uniquely Mapping Ratio |
|---|---|---|---|---|---|---|
| Wei_1 | SRR5577192 | 65911108 | 52272948 | 27513396 | 96.78% | 52.60% |
| Wei_2 | SRR5577193 | 65914286 | 53989652 | 29869086 | 95.69% | 55.30% |
| Wei_3 | SRR5577194 | 93927314 | 75245286 | 39681060 | 96.16% | 52.70% |
| Yor_1 | SRR5577189 | 104766230 | 84176946 | 46670672 | 96.78% | 55.40% |
| Yor_2 | SRR5577190 | 72593892 | 58591238 | 32183620 | 96.84% | 54.90% |
| Yor_3 | SRR5577191 | 105590048 | 86535990 | 49194174 | 96.80% | 56.80% |
| DEL | Adjacent Protein-Coding Gene | Pearson Correlation Coefficient | p-Value |
|---|---|---|---|
| MSTRG.12725 | ENSSSCG00000002469(OTUB2) | −0.915706465 | 0.010358 |
| ENSSSCG00000039415(CCDC19) | 0.817455545 | 0.04694 | |
| MSTRG.13894 | ENSSSCG00000039986(RGS8) | 0.950415666 | 0.003626 |
| MSTRG.2101 | ENSSSCG00000037202(CACNG4) | 0.826596447 | 0.042496 |
| MSTRG.3671 | ENSSSCG00000038948(ETS) | 0.87248832 | 0.02335 |
| MSTRG.4937 | ENSSSCG00000015981(HOXD10) | 0.939146854 | 0.00544 |
| ENSSSCG00000015986(HOXD1) | 0.919787616 | 0.00939 | |
| ENSSSCG00000034741(HOXD11) | 0.840680905 | 0.03605 | |
| MSTRG.8326 | ENSSSCG00000008218(RNF103) | 0.86271199 | 0.026978 |
| ENSSSCG00000035478(RMND5A) | 0.929580341 | 0.00726 | |
| MSTRG.8829 | ENSSSCG00000005970(SQLE) | 0.965516179 | 0.001763 |
| DE lincRNAs | Number | DE lincRNAs | Number | ||||
|---|---|---|---|---|---|---|---|
| DEPTGs | UpRegulated PTGs | DownRegulated PTGs | DEPTGs | UpRegulated PTGs | DownRegulated PTGs | ||
| MSTRG.10534 | 86 | 58 | 28 | MSTRG.3619 | 32 | 32 | 0 |
| MSTRG.11176 | 13 | 13 | 0 | MSTRG.4175 | 40 | 40 | 0 |
| MSTRG.12725 | 14 | 12 | 2 | MSTRG.4937 | 47 | 47 | 0 |
| MSTRG.1306 | 77 | 27 | 50 | MSTRG.5833 | 57 | 49 | 8 |
| MSTRG.13894 | 68 | 66 | 2 | MSTRG.6103 | 82 | 70 | 12 |
| MSTRG.2101 | 12 | 11 | 1 | MSTRG.62 | 25 | 24 | 1 |
| MSTRG.3426 | 32 | 30 | 0 | MSTRG.8326 | 54 | 48 | 6 |
| MSTRG.3546 | 64 | 64 | 0 | MSTRG.8829 | 10 | 10 | 0 |
| MSTRG.130 | 4 | 4 | 0 | MSTRG.3671 | 20 | 20 | 0 |
| MSTRG.13805 | 19 | 19 | 0 | MSTRG.4329 | 20 | 20 | 0 |
| MSTRG.13909 | 5 | 5 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Huang, Z.; Zhao, W.; Li, M.; Li, C. Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. Int. J. Mol. Sci. 2020, 21, 1732. https://doi.org/10.3390/ijms21051732
Li Q, Huang Z, Zhao W, Li M, Li C. Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. International Journal of Molecular Sciences. 2020; 21(5):1732. https://doi.org/10.3390/ijms21051732
Chicago/Turabian StyleLi, Qianqian, Ziying Huang, Wenjuan Zhao, Mengxun Li, and Changchun Li. 2020. "Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs" International Journal of Molecular Sciences 21, no. 5: 1732. https://doi.org/10.3390/ijms21051732
APA StyleLi, Q., Huang, Z., Zhao, W., Li, M., & Li, C. (2020). Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. International Journal of Molecular Sciences, 21(5), 1732. https://doi.org/10.3390/ijms21051732