Excess Accumulation of Lipid Impairs Insulin Sensitivity in Skeletal Muscle

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Insulin Signaling Pathway and Glucose Uptake in Skeletal Muscle
3. High Fat Accumulation Triggers Insulin Resistance in Skeletal Muscle
4. Maintaining Cell Homeostasis in Skeletal Muscle
5. Exercise Effects on Glucose Uptake in Muscle Insulin Resistance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Kahn, R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Honka, M.-J.; Latva-Rasku, A.; Bucci, M.; Virtanen, K.A.; Hannukainen, J.C.; Kalliokoski, K.K.; Nuutila, P. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: A positron emission tomography study. Eur. J. Endocrinol. 2018, 178, 523–531. [Google Scholar] [CrossRef]
- Klip, A.; Paquet, M.R. Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care 1990, 13, 228–243. [Google Scholar] [CrossRef]
- Cartee, G.D. Mechanisms for greater insulin-stimulated glucose uptake in normal and insulin-resistant skeletal muscle after acute exercise. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E949–E959. [Google Scholar] [CrossRef] [Green Version]
- Merry, T.L.; McConell, G.K. Skeletal muscle glucose uptake during exercise: A focus on reactive oxygen species and nitric oxide signaling. IUBMB Life 2009, 61, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbarino, J.; Sturley, S.L. Saturated with fat: New perspectives on lipotoxicity. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Neess, D.; Bek, S.; Engelsby, H.; Gallego, S.F.; Færgeman, N.J. Long-chain acyl-CoA esters in metabolism and signaling: Role of acyl-CoA binding proteins. Prog. Lipid Res. 2015, 59, 1–25. [Google Scholar] [CrossRef]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Lu, A.M.; Wang, Y.; Hong, A.; Chen, Y.; Hu, J.; Li, X.; Qin, Z.H. Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp. Gerontol. 2013, 48, 427–436. [Google Scholar] [CrossRef]
- Kennedy, J.W.; Hirshman, M.F.; Gervino, E.V.; Ocel, J.V.; Forse, R.A.; Hoenig, S.J.; Aronson, D.; Goodyear, L.J.; Horton, E.S. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 1999, 48, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Rodnick, K.J.; Slot, J.W.; Studelska, D.R.; Hanpeter, D.E.; Robinson, L.J.; Geuze, H.J.; James, D.E. Immunocytochemical and biochemical studies of GLUT4 in rat skeletal muscle. J. Biol. Chem. 1992, 267, 6278–6285. [Google Scholar] [PubMed]
- Taylor, E.B.; An, D.; Kramer, H.F.; Yu, H.; Fujii, N.L.; Roeckl, K.S.C.; Bowles, N.; Hirshman, M.F.; Xie, J.; Feener, E.P.; et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J. Biol. Chem. 2008, 283, 9787–9796. [Google Scholar] [CrossRef] [Green Version]
- Bettedi, L.; Foukas, L.C. Growth factor, energy and nutrient sensing signalling pathways in metabolic ageing. Biogerontology 2017, 18, 913–992. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–921. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.B.; Zheng, X.; Agrawal, S.; Cartee, G.D. Whole body glucoregulation and tissue-specific glucose uptake in a novel Akt substrate of 160 kDa knockout rat model. PLoS ONE 2019, 14, e0216236. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake—regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Jaiswal, N.; Gavin, M.G.; Quinn, W.J.; Luongo, T.S.; Gelfer, R.G.; Baur, J.A.; Titchenell, P.M. The role of skeletal muscle Akt in the regulation of muscle mass and glucose homeostasis. Mol. Metab. 2019, 28, 1–13. [Google Scholar] [CrossRef]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2014, 26, 323–331. [Google Scholar] [CrossRef]
- Takenaka, N.; Araki, N.; Satoh, T. Involvement of the protein kinase Akt2 in insulin-stimulated Rac1 activation leading to glucose uptake in mouse skeletal muscle. PLoS ONE 2019, 14, e0212219. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Krause, U.; Desmondt, F.; Tanti, J.-F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ijuin, T.; Takenawa, T. Regulation of insulin signaling and glucose transporter 4 (GLUT4) exocytosis by phosphatidylinositol 3,4,5-trisphosphate (PIP3) phosphatase, skeletal muscle, and kidney enriched inositol polyphosphate phosphatase (SKIP). J. Biol. Chem. 2012, 287, 6991–6999. [Google Scholar] [CrossRef] [Green Version]
- Ijuin, T.; Takenawa, T. Role of phosphatidylinositol 3,4,5-trisphosphate (PIP3) 5-phosphatase skeletal muscle- and kidney-enriched inositol polyphosphate phosphatase (SKIP) in myoblast differentiation. J. Biol. Chem. 2012, 287, 31330–31341. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; Jenkinson, C.P.; Richardson, D.K.; Tripathy, D.; DeFronzo, R.A. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: Results from the Veterans Administration Genetic Epidemiology Study. Diabetes 2006, 55, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Krook, A.; Bjornholm, M.; Galuska, D.; Jiang, X.J.; Fahlman, R.; Myers, M.G., Jr.; Wallberg-Henriksson, H.; Zierath, J.R. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Balas, B.; Christ-Roberts, C.Y.; Kim, R.Y.; Ramos, F.J.; Kikani, C.K.; Li, C.; Deng, C.; Reyna, S.; Musi, N.; et al. Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Mol. Cell. Biol. 2007, 27, 6497–6505. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Ducommun, S.; Quan, C.; Xie, B.; Li, M.; Wasserman, D.H.; Sakamoto, K.; Mackintosh, C.; Chen, S. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. Biochem. J. 2013, 449, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [Green Version]
- Morignyab, P.; Houssierab, M.; Mouiselab, E.; Langina, D. Adipocyte lipolysis and insulin resistance. Biochmie 2016, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Gunilla, O. Role of lipoprotein lipase in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar]
- Parafati, M.; Lascala, A.; Morittu, V.M.; Trimboli, F.; Rizzuto, A.; Brunelli, E.; Coscarelli, F.; Costa, N.; Britti, D.; Ehrlich, J.; et al. Bergamot polyphenol fraction prevents nonalcoholic fatty liver disease via stimulation of lipophagy in cafeteria diet-induced rat model of metabolic syndrome. J. Nutr. Biochem. 2015, 26, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castro, C.; Garvey, W.T. Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 841–856. [Google Scholar] [CrossRef] [Green Version]
- Kamei, Y.; Miura, S.; Suganami, T.; Akaike, F.; Kanai, S.; Sugita, S.; Katsumata, A.; Aburatani, H.; Unterman, T.G.; Ezaki, O.; et al. Regulation of SREBP1c gene expression in skeletal muscle: Role of Retinoid X Receptor/Liver X Receptor and Forkhead-O1 Transcription Factor. Endocrinology 2008, 149, 2293–2305. [Google Scholar] [CrossRef] [Green Version]
- Guillet-Deniau, I.; Mieulet, V.; le Lay, S.; Achouri, Y.; Carre, D.; Girard, J.; Foufelle, F.; Ferre, P. Sterol regulatory element binding protein-1c expression and action in rat muscles: Insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes 2002, 51, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Guillet-Deniau, I.; Pichard, A.L.; Kone, A.; Esnous, C.; Nieruchalski, M.; Girard, J.; Prip-Buus, C. Glucose induces de novo lipogenesis in rat muscle satellite cells through a sterol-regulatory-element-binding-protein-1c-dependent pathway. J. Cell Sci. 2004, 117, 1937–1944. [Google Scholar] [CrossRef] [Green Version]
- Lecomte, V.; Meugnier, E.; Euthine, V.; Durand, C.; Freyssenet, D. A new role for sterol regulatory element binding protein 1 transcription factors in the regulation of muscle mass and muscle cell differentiation. Mol. Cell. Biol. 2010, 30, 1182–1198. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Sung, C.K.; Abbott, M.J.; Turcotte, L.P. Phosphatidylinositol 3-kinase-dependent insulin regulation of long-chain fatty acid (LCFA) metabolism in L6 muscle cells: Involvement of atypical protein kinase Czeta in LCFA uptake but not oxidation. J. Endocrinol. 2008, 198, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Dessalle, K.; Euthine, V.; Chanon, S.; Delarichaudy, J.; Fujii, I.; Rome, S.; Vidal, H.; Nemoz, G.; Simon, C.; Lefai, E. SREBP-1 Transcription Factors Regulate Skeletal Muscle Cell Size by Controlling Protein Synthesis through Myogenic Regulatory Factors. PLoS ONE 2012, 17, e50878. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Bezy, O.; Tran, T.T.; Pihlajamaki, J.; Suzuki, R.; Emanuelli, B.; Winnay, J.; Mori, M.A.; Haas, J.; Biddinger, S.B.; Leitges, M.; et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Investig. 2011, 121, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Soundarapandian, M.M.; Sessions, H.; Peddibhotla, S.; Roth, G.P.; Li, J.L.; Sugarman, E.; Koo, A.; Malany, S.; Wang, M.; et al. MondoA coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J. Clin. Investig. 2016, 126, 3567–3579. [Google Scholar] [CrossRef]
- Ahn, B.; Wan, S.; Jaiswal, N.; Vega, R.B.; Ayer, D.E.; Titchenell, P.M.; Han, X.; Won, K.J.; Kelly, D.P. MondoA drives muscle lipid accumulation and insulin resistance. JCI Insight 2019, 4, e129119. [Google Scholar] [CrossRef]
- Song, Z.; Yang, H.; Zhou, L.; Yang, F. Glucose-sensing transcription factor mondoa/ChREBP as targets for type 2 diabetes: Opportunities and challenges. Int. J. Mol. Sci. 2019, 20, 5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Morris, K.J.; Ng, Y.C. Fiber type-specific immunostaining of the Na+, K+-ATPase subunit isoforms in skeletal muscle: Age-associated differential changes. Biochim. Biophys. Acta 2006, 1762, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Albers, P.H.; Pedersen, A.J.; Birk, J.B.; Kristensen, D.E.; Vind, B.F.; Baba, O.; Nohr, J.; Hojlund, K.; Wojtaszewski, J.F. Human muscle fiber type-specific insulin signaling: Impact of obesity and type 2 diabetes. Diabetes 2015, 64, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Funai, K.; Lodhi, I.J.; Spears, L.D.; Yin, L.; Song, H.; Klein, S.; Semenkovich, C.F. Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes 2016, 65, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, R.A.; Sepa-Kishi, D.M.; Bikopoulos, G.; Wu, M.V.; Uthayakumar, A.; Mohasses, A.; Hughes, M.C.; Perry, C.G.R.; Ceddia, R.B. High-fat diet induces skeletal muscle oxidative stress in a fiber type-dependent manner in rats. Free Radic. Biol. Med. 2017, 110, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Ourabah, S.; Montagne, J.; Burnol, A.F.; Postic, C.; Guilmeau, S. MondoA/ChREBP: The usual suspects of transcriptional glucose sensing; Implication in pathophysiology. Metabolism 2017, 70, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Dyck, D.J.; Luiken, J.J. Skeletal muscle fatty acid transport and transporters. Adv. Exp. Med. Biol. 1998, 441, 193–205. [Google Scholar]
- Glatz, J.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef]
- Ko, K.; Woo, J.; Bae, J.Y.; Roh, H.T.; Lee, Y.H.; Shin, K.O. Exercise training improves intramuscular triglyceride lipolysis sensitivity in high-fat diet induced obese mice. Lipids Health Dis. 2018, 17, 81. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Grunfeld, C. Introduction to Lipids and Lipoproteins. In Endotext [Internet]; MDText. com Inc. U.S. National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- Li, Y.; Xu, S.; Zhang, X.; Yi, Z.; Cichello, S. Skeletal intramyocellular lipid metabolism and insulin resistance. Biophys. Rep. 2015, 1, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of lipid droplets and perilipins. J. Diabetes Res. 2017, 2017, 1789395. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-induced insulin resistance in skeletal muscle: The chase for the culprit goes from total intramuscular fat to lipid intermediates, and finally to species of lipid intermediates. Nutrients 2016, 8, 466. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [PubMed] [Green Version]
- Glatz, J.; Luiken, J.J.; Bonen, A. Membrance fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Tepaamorndech, S.; Kirschke, C.P.; Newman, J.W.; Keyes, W.R.; Pedersen, T.L.; Dumnil, J. Aberrant fatty acid metabolism in skeletal muscle contributes to insulin resistance in zinc transporter 7 (znt7)-knockout mice. J. Biol. Chem. 2018, 293, 7549–7563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (dys)function and insulin resistance: From pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annual Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Craessaerts, K.; Strooper, B.D.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, N.B.; Dorn, G.W.; Yin, X.-M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.; Xu, Z.; Liu, L.; Guo, Q.; Wu, H.; Liang, X.; Zhou, D.; Xiao, L.; Liu, L.; Liu, Y.; et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 2018, 23, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Motiani, P.; Virtanen, K.A.; Motiani, K.K.; Eskelinen, J.J.; Middelbeek, R.J.; Goodyear, L.J.; Savolainen, A.M.; Kemppainen, J.; Jensen, J.; Din, M.U.; et al. Decreased insulin-stimulated brown adipose tissue glucose uptake after short-term exercise training in healthy middle-aged men. Diabetes Obes. Metab. 2017, 19, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, P.; Krotkiewski, M.; Holm, G.; Gustafsson, C.; Bjorntorp, P. Effects of acute exercise on insulin and non-insulin-dependent glucose uptake in normal and moderately obese women. Eur. J. Med. 1993, 2, 199–204. [Google Scholar] [PubMed]
- Jensen, T.E.; Maarbjerg, S.J.; Rose, A.J.; Leitges, M.; Richter, E.A. Knockout of the predominant conventional PKC isoform, PKCalpha, in mouse skeletal muscle does not affect contraction-stimulated glucose uptake. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E340–E348. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hayashi, T.; Toyoda, T.; Hamada, T.; Shimizu, Y.; Hirata, M.; Ebihara, K.; Masuzaki, H.; Hosoda, K.; Fushiki, T.; et al. High-fat diet impairs the effects of a single bout of endurance exercise on glucose transport and insulin sensitivity in rat skeletal muscle. Metabolism 2007, 56, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Way, K.L.; Hackett, D.A.; Baker, M.K.; Johnson, N.A. The effect of regular exercise on insulin sensitivity in type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Metab. J. 2016, 40, 253–271. [Google Scholar]
- Sharma, P.; Arias, E.B.; Cartee, G.D. Protein phosphatase 1-alpha regulates AS160 Ser588 and Thr642 dephosphorylation in skeletal muscle. Diabetes 2016, 65, 2606–2617. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.B.; Wang, H.; Cartee, G.D. Akt substrate of 160 kDa dephosphorylation rate is reduced in insulin-stimulated rat skeletal muscle after acute exercise. Physiol. Res. 2018, 67, 143–147. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kuramoto, K.; Wang, N.; Situ, X.; Priyadarshini, M.; Zhang, W.; Cordoba-Chacon, J.; Layden, B.T.; He, C. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 2018, 23, 3286–3299. [Google Scholar] [CrossRef]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, I.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y.; Yeom, D.C. Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis. J. Exerc. Nutr. Biochem. 2015, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, S.; Langleite, T.; Norheim, F.; Pourteymour, S.; Jensen, J.; Stadheim, H.K.; Storas, T.H.; Davanger, S.; Gulseth, H.L. Subsarcolemmal lipid droplet responses to a combined endurance and strength exercise intervention. Physiol. Rep. 2014, 2, e12187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, S.; Gemmink, A.; Brouwers, B.; Meex, R.C.R.; Huntjens, P.R.; Schaart, G.; Moonen-Kornips, E.; Jorgensen, J.; Hoeks, J.; Schrauwen, P. Distinct lipid droplet characteristics and distribution unmask the apparent contradiction of the athlete’s paradox. Mol. Metab. 2018, 17, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Marcinko, K.; Bujak, A.L.; Lally, J.S.; Ford, R.J.; Wong, T.H.; Smith, B.K.; Kemp, B.E.; Jenkins, Y.; Li, W.; Kinsella, T.M.; et al. The AMPK activator R419 improves exercise capacity and skeletal muscle insulin sensitivity in obese mice. Mol. Metab. 2015, 4, 643–651. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M. AMPK and exercise: Glucose uptake and insulin sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Kjobsted, R.; Munk-Hansen, N.; Birk, J.B.; Foretz, M.; Viollet, B.; Bjornholm, M.; Zierath, J.R.; Treebak, J.T.; Wojtaszewski, J.F. Enhanced Muscle Insulin Sensitivity After Contraction/Exercise Is Mediated by AMPK. Diabetes 2017, 66, 598–612. [Google Scholar] [CrossRef] [Green Version]
- Zaid, H.; Talior-Volodarsky, I.; Antonescu, C.; Liu, Z.; Klip, A. GAPDH binds GLUT4 reciprocally to hexokinase-II and regulates glucose transport activity. Biochem. J. 2009, 419, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.J.; Pratipanawatr, T.; Koval, J.; Printz, R.; Ardehali, H.; Granner, D.K.; Defronzo, R.A.; Mandarino, L.J. Exercise increases hexokinase II mRNA, but not activity in obesity and type 2 diabetes. Metabolism 2001, 50, 602–606. [Google Scholar] [CrossRef]
- Pedersen, A.J.; Hingst, J.R.; Friedrichsen, M.; Kristensen, J.M.; Hojlund, K.; Wojtaszewski, J.F. Dysregulation of muscle glycogen synthase in recovery from exercise in type 2 diabetes. Diabetologia 2015, 58, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.; Stepto, N.K.; Shaw, C.S.; Serpiello, F.R.; Anderson, M.; Hare, D.L.; Levinger, I. Acute High-intensity interval exercise-induced redox signaling is associated with enhanced insulin sensitivity in obese middle-aged men. Front. Physiol. 2016, 7, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa-Caldwell, M.E.; Brown, J.L.; Lee, D.E.; Blackwell, T.A.; Turner, K.W.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Washington, T.A.; Greene, N.P. Autophagy activation, not peroxisome proliferator-activated receptor gamma coactivator 1alpha, may mediate exercise-induced improvements in glucose handling during diet-induced obesity. Exp. Physiol. 2017, 102, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- La Fuente, F.P.; Quezada, L.; Sepulveda, C.; Monsalves-Alvarez, M.; Rodriguez, J.M.; Sacristan, C.; Chiong, M.; Llanos, M.; Espinosa, A.; Troncoso, R. Exercise regulates lipid droplet dynamics in normal and fatty liver. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2019, 1864, 158519. [Google Scholar] [CrossRef]

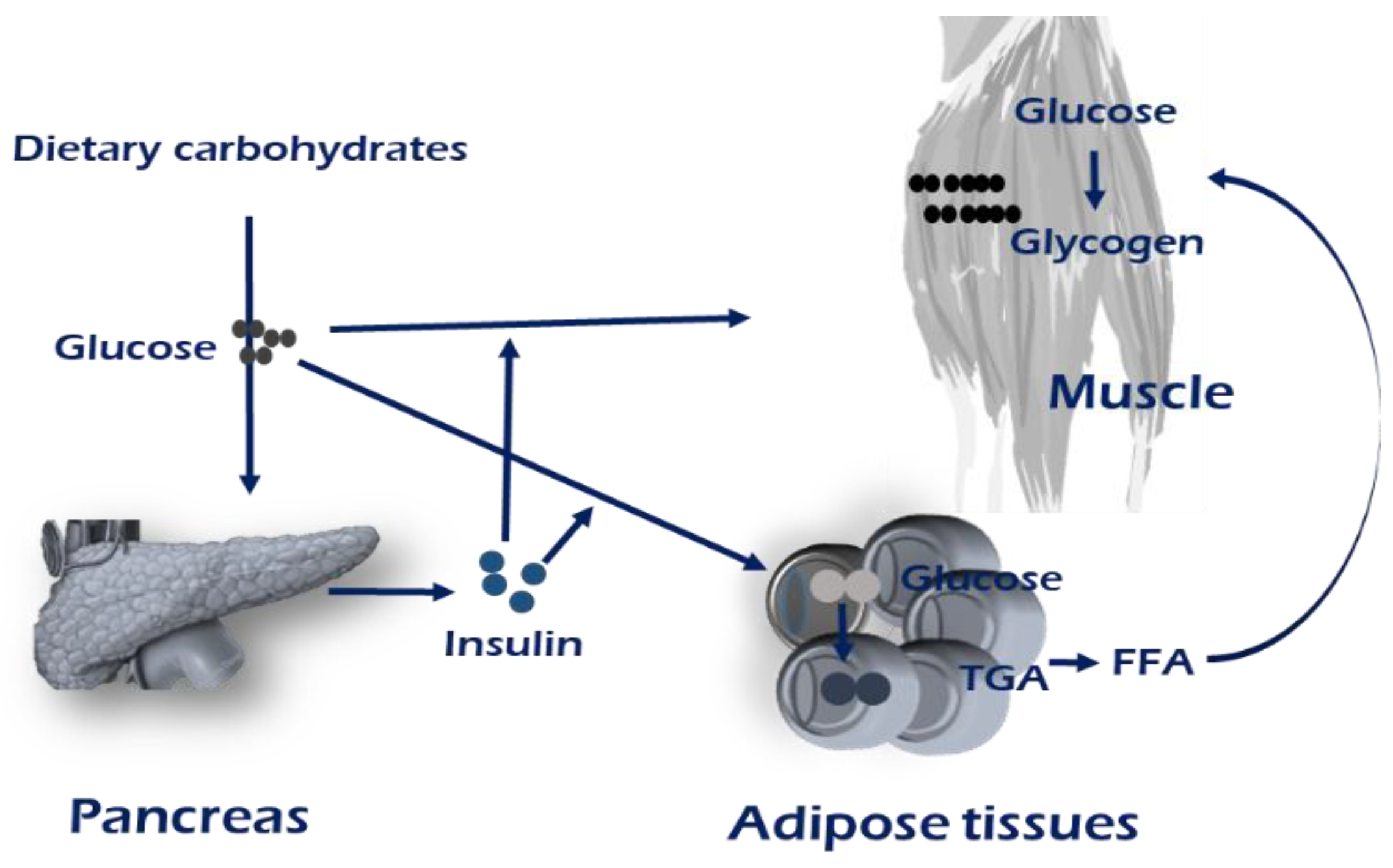
 Insulin receptor,
Insulin receptor,  GLUT4,
GLUT4,  CD36.
Insulin receptor, GLUT4, CD36.
CD36.
Insulin receptor, GLUT4, CD36.
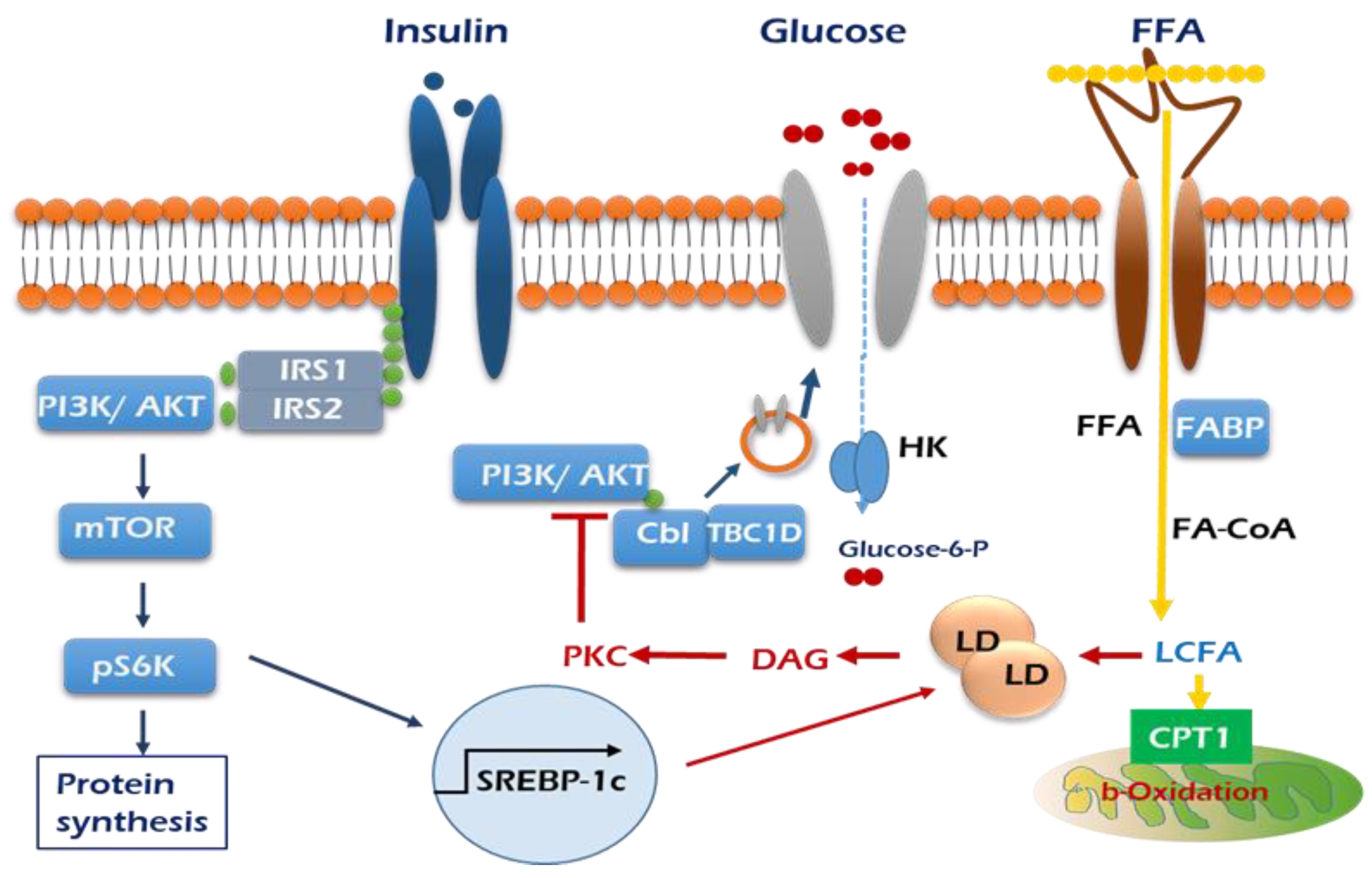
 , GLUT4; HK, hexokinase.
, GLUT4; HK, hexokinase.
, GLUT4; HK, hexokinase.
, GLUT4; HK, hexokinase.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.S.; Seo, Y.-K. Excess Accumulation of Lipid Impairs Insulin Sensitivity in Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 1949. https://doi.org/10.3390/ijms21061949
Park SS, Seo Y-K. Excess Accumulation of Lipid Impairs Insulin Sensitivity in Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(6):1949. https://doi.org/10.3390/ijms21061949
Chicago/Turabian StylePark, Sung Sup, and Young-Kyo Seo. 2020. "Excess Accumulation of Lipid Impairs Insulin Sensitivity in Skeletal Muscle" International Journal of Molecular Sciences 21, no. 6: 1949. https://doi.org/10.3390/ijms21061949
APA StylePark, S. S., & Seo, Y. -K. (2020). Excess Accumulation of Lipid Impairs Insulin Sensitivity in Skeletal Muscle. International Journal of Molecular Sciences, 21(6), 1949. https://doi.org/10.3390/ijms21061949