Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease


Abstract
:1. Introduction
2. Vasoactive Pathways
2.1. RAAS Pathway
2.1.1. Renin-Angiotensin-Aldosterone System (RAAS)
2.1.2. Vasodilatory Arm of RAAS
2.1.3. The RAAS Inhibition as a Therapeutic Target
2.2. Endothelins
2.3. Urotensin II
3. Metabolic Pathways
3.1. Mitochondria and Reactive Oxygen Species (ROS)
3.2. NADPH Oxidase (NOX)
3.3. Nitric Oxide Synthase
3.4. Dicarbonyl Synthesis
MGO as a Therapeutic Target
3.5. Advanced Glycation and RAGE
4. Hemodynamic and Metabolic Pathway Interactions
5. New Targets for Renoprotection
5.1. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors
5.2. Incretin-Related Therapies
5.2.1. GLP-1 Receptor Agonists
5.2.2. DPP-4 Inhibitors
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathis, D.; Vence, L.; Benoist, C. Beta-Cell death during progression to diabetes. Nature 2001, 414, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001, 44, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.R.; Dzau, V.J. The renin-angiotensin-aldosterone system: A specific target for hypertension management. Am. J. Hypertens. 1999, 12, 205S–213S. [Google Scholar] [CrossRef]
- Navar, L.G.; Imig, J.D.; Zou, L.; Wang, C.T. Intrarenal production of angiotensin II. Semin. Nephrol. 1997, 17, 412–422. [Google Scholar]
- Yoo, T.H.; Li, J.J.; Kim, J.J.; Jung, D.S.; Kwak, S.J.; Ryu, D.R.; Choi, H.Y.; Kim, J.S.; Kim, H.J.; Han, S.H.; et al. Activation of the renin-angiotensin system within podocytes in diabetes. Kidney Int. 2007, 71, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Noble, N.A.; Zhang, J.; Xu, C.; Border, W.A. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007, 72, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, S.B.; Mauer, M.; Basgen, J.M.; Aguiniga, E.; Chon, Y. Effect of angiotensin II on glomerular structure in streptozotocin-induced diabetic rats. Am. J. Nephrol. 2004, 24, 549–556. [Google Scholar] [CrossRef]
- Tone, A.; Shikata, K.; Ogawa, D.; Sasaki, S.; Nagase, R.; Sasaki, M.; Yozai, K.; Usui, H.K.; Okada, S.; Wada, J.; et al. Changes of gene expression profiles in macrophages stimulated by angiotensin II--angiotensin II induces MCP-2 through AT1-receptor. J. Renin Angiotensin Aldosterone Syst. 2007, 8, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelfinger, J.R.; Zuo, W.M.; Fon, E.A.; Ellison, K.E.; Dzau, V.J. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J. Clin. Investig. 1990, 85, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, Y.; Tomita, K.; Nonoguchi, H.; Marumo, F. PCR localization of angiotensin II receptor and angiotensinogen mRNAs in rat kidney. Kidney Int. 1993, 43, 1251–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Chen, S.; Chiu, J.; George, B.; Chakrabarti, S. Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1119–E1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric, C. Vasoactive hormones and the diabetic kidney. Sci World J. 2008, 8, 470–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.M.; Knudsen, S.T.; Cooper, M.E. Diabetic nephropathy: An insight into molecular mechanisms and emerging therapies. Expert Opin. Ther. Targets 2019, 23, 579–591. [Google Scholar] [CrossRef]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press. 2003, 12, 70–88. [Google Scholar] [CrossRef]
- Higuchi, S.; Ohtsu, H.; Suzuki, H.; Shirai, H.; Frank, G.D.; Eguchi, S. Angiotensin II signal transduction through the AT1 receptor: Novel insights into mechanisms and pathophysiology. Clin. Sci (Lond.) 2007, 112, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Wichi, R.B.; Farah, V.; Chen, Y.; Irigoyen, M.C.; Morris, M. Deficiency in angiotensin AT1a receptors prevents diabetes-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1184–R1189. [Google Scholar] [CrossRef] [Green Version]
- Langham, R.G.; Kelly, D.J.; Cox, A.J.; Thomson, N.M.; Holthofer, H.; Zaoui, P.; Pinel, N.; Cordonnier, D.J.; Gilbert, R.E. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: Effects of angiotensin converting enzyme inhibition. Diabetologia 2002, 45, 1572–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gasparo, M.; Husain, A.; Alexander, W.; Catt, K.J.; Chiu, A.T.; Drew, M.; Goodfriend, T.; Harding, J.W.; Inagami, T.; Timmermans, P.B. Proposed update of angiotensin receptor nomenclature. Hypertension 1995, 25, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Morley, A.L.; Koitka, A.; Samuel, P.; Coughlan, M.T.; Penfold, S.A.; Thomas, M.C.; Bierhaus, A.; Nawroth, P.P.; Yamamoto, H.; et al. Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Diabetologia 2010, 53, 2442–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria-Costa, G.; Leite-Moreira, A.; Henriques-Coelho, T. Cardiovascular effects of the angiotensin type 2 receptor. Rev. Port. Cardiol. 2014, 33, 439–449. [Google Scholar] [CrossRef]
- Koitka, A.; Cao, Z.; Koh, P.; Watson, A.M.; Sourris, K.C.; Loufrani, L.; Soro-Paavonen, A.; Walther, T.; Woollard, K.J.; Jandeleit-Dahm, K.A.; et al. Angiotensin II subtype 2 receptor blockade and deficiency attenuate the development of atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes. Diabetologia 2010, 53, 584–592. [Google Scholar] [CrossRef]
- Brassard, P.; Amiri, F.; Thibault, G.; Schiffrin, E.L. Role of angiotensin type-1 and angiotensin type-2 receptors in the expression of vascular integrins in angiotensin II-infused rats. Hypertension 2006, 47, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Dandapat, A.; Hu, C.P.; Chen, J.; Liu, Y.; Khan, J.A.; Remeo, F.; Carey, R.M.; Hermonat, P.L.; Mehta, J.L. Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem. Biophys. Res. Commun. 2008, 366, 871–877. [Google Scholar] [CrossRef]
- Hu, C.; Dandapat, A.; Chen, J.; Liu, Y.; Hermonat, P.L.; Carey, R.M.; Mehta, J.L. Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis 2008, 199, 288–294. [Google Scholar] [CrossRef]
- Savoia, C.; Ebrahimian, T.; He, Y.; Gratton, J.P.; Schiffrin, E.L.; Touyz, R.M. Angiotensin II/AT2 receptor-induced vasodilation in stroke-prone spontaneously hypertensive rats involves nitric oxide and cGMP-dependent protein kinase. J. Hypertens. 2006, 24, 2417–2422. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. The role of aldosterone in the metabolic syndrome. Curr. Hypertens. Rep. 2011, 13, 163–172. [Google Scholar] [CrossRef]
- Sato, A.; Saruta, T. Aldosterone breakthrough during angiotensin-converting enzyme inhibitor therapy. Am. J. Hypertens. 2003, 16, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Schjoedt, K.J.; Rossing, K.; Juhl, T.R.; Boomsma, F.; Tarnow, L.; Rossing, P.; Parving, H.H. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006, 70, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.Y.; Kim, C.H.; Kim, H.S.; Jee, Y.H.; Song, H.K.; Lee, M.H.; Han, K.H.; Kim, H.K.; Kang, Y.S.; Han, J.Y.; et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J. Am. Soc. Nephrol. 2006, 17, 1362–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, M.; Williams, G.H.; Weinberger, M.; Lewin, A.; Krause, S.; Mukherjee, R.; Patni, R.; Beckerman, B. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin. J. Am. Soc. Nephrol. 2006, 1, 940–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakris, G.L.; Agarwal, R.; Chan, J.C.; Cooper, M.E.; Gansevoort, R.T.; Haller, H.; Remuzzi, G.; Rossing, P.; Schmieder, R.E.; Nowack, C.; et al. Effect of Finerenone on Albuminuria in Patients With Diabetic Nephropathy: A Randomized Clinical Trial. JAMA 2015, 314, 884–894. [Google Scholar] [CrossRef]
- Mendoza-Torres, E.; Oyarzun, A.; Mondaca-Ruff, D.; Azocar, A.; Castro, P.F.; Jalil, J.E.; Chiong, M.; Lavandero, S.; Ocaranza, M.P. ACE2 and vasoactive peptides: Novel players in cardiovascular/renal remodeling and hypertension. Ther. Adv. Cardiovasc. Dis. 2015, 9, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Michea, L.; Chiong, M.; Lagos, C.F.; Lavandero, S.; Jalil, J.E. Recent insights and therapeutic perspectives of angiotensin-(1-9) in the cardiovascular system. Clin. Sci. (Lond.) 2014, 127, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Chamsi-Pasha, M.A.; Shao, Z.; Tang, W.H. Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr. Heart Fail. Rep. 2014, 11, 58–63. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Tikellis, C.; Johnston, C.I.; Forbes, J.M.; Burns, W.C.; Burrell, L.M.; Risvanis, J.; Cooper, M.E. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension 2003, 41, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M. Angiotension-(1-7) and antihypertensive mechanisms. J. Nephrol. 1998, 11, 278–283. [Google Scholar] [PubMed]
- Trask, A.J.; Ferrario, C.M. Angiotensin-(1-7): Pharmacology and new perspectives in cardiovascular treatments. Cardiovasc. Drug Rev. 2007, 25, 162–174. [Google Scholar] [CrossRef]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.D. The emerging role of angiotensin-converting enzyme-2 in the kidney. Curr. Opin. Nephrol. Hypertens. 2007, 16, 116–121. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Iyer, S.N. Angiotensin-(1-7): A bioactive fragment of the renin-angiotensin system. Regul. Pept. 1998, 78, 13–18. [Google Scholar] [CrossRef]
- Benter, I.F.; Yousif, M.H.; Anim, J.T.; Cojocel, C.; Diz, D.I. Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H684–H691. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tadano, T.; Tan-No, K. Angiotensin (1-7) prevents angiotensin II-induced nociceptive behaviour via inhibition of p38 MAPK phosphorylation mediated through spinal Mas receptors in mice. Eur. J. Pain 2014, 18, 1471–1479. [Google Scholar] [CrossRef]
- Zheng, J.; Li, G.; Chen, S.; Bihl, J.; Buck, J.; Zhu, Y.; Xia, H.; Lazartigues, E.; Chen, Y.; Olson, J.E. Activation of the ACE2/Ang-(1-7)/Mas pathway reduces oxygen-glucose deprivation-induced tissue swelling, ROS production, and cell death in mouse brain with angiotensin II overproduction. Neuroscience 2014, 273, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Drummer, O.H.; Kourtis, S.; Johnson, H. Effect of chronic enalapril treatment on enzymes responsible for the catabolism of angiotensin I and formation of angiotensin II. Biochem. Pharmacol. 1990, 39, 513–518. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Godoy, I.; Jalil, J.E.; Varas, M.; Collantes, P.; Pinto, M.; Roman, M.; Ramirez, C.; Copaja, M.; Diaz-Araya, G.; et al. Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006, 48, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Ocaranza, M.P.; Lavandero, S.; Jalil, J.E.; Moya, J.; Pinto, M.; Novoa, U.; Apablaza, F.; Gonzalez, L.; Hernandez, C.; Varas, M.; et al. Angiotensin-(1-9) regulates cardiac hypertrophy in vivo and in vitro. J. Hypertens. 2010, 28, 1054–1064. [Google Scholar] [CrossRef]
- Flores-Munoz, M.; Godinho, B.M.; Almalik, A.; Nicklin, S.A. Adenoviral delivery of angiotensin-(1-7) or angiotensin-(1-9) inhibits cardiomyocyte hypertrophy via the mas or angiotensin type 2 receptor. PLoS ONE 2012, 7, e45564. [Google Scholar] [CrossRef]
- Cha, S.A.; Park, B.M.; Gao, S.; Kim, S.H. Stimulation of ANP by angiotensin-(1-9) via the angiotensin type 2 receptor. Life Sci. 2013, 93, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Rivera, P.; Novoa, U.; Pinto, M.; Gonzalez, L.; Chiong, M.; Lavandero, S.; Jalil, J.E. Rho kinase inhibition activates the homologous angiotensin-converting enzyme-angiotensin-(1-9) axis in experimental hypertension. J. Hypertens. 2011, 29, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Munoz, M.; Work, L.M.; Douglas, K.; Denby, L.; Dominiczak, A.F.; Graham, D.; Nicklin, S.A. Angiotensin-(1-9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 2012, 59, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parving, H.H.; Lehnert, H.; Brochner-Mortensen, J.; Gomis, R.; Andersen, S.; Arner, P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N. Engl. J. Med. 2001, 345, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S105–S118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Pressel, S.; Davis, B.R.; Nwachuku, C.; Wright, J.T., Jr.; Whelton, P.K.; Barzilay, J.; Batuman, V.; Eckfeldt, J.H.; Farber, M.; et al. Renal outcomes in high-risk hypertensive patients treated with an angiotensin-converting enzyme inhibitor or a calcium channel blocker vs a diuretic: A report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch. Intern. Med. 2005, 165, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Group, A.C.; MacMahon, S.; Chalmers, J.; Neal, B.; Woodward, M.; Billot, L.; Harrap, S.; Poulter, N.; Marre, M.; et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): A randomised controlled trial. Lancet 2007, 370, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Ford, C.E.; Cutler, J.A.; Davis, B.R.; Piller, L.B.; Whelton, P.K.; Wright, J.T., Jr.; Barzilay, J.I.; Brown, C.D.; Colon, P.J., Sr.; et al. Long-term renal and cardiovascular outcomes in Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) participants by baseline estimated GFR. Clin. J. Am. Soc. Nephrol. 2012, 7, 989–1002. [Google Scholar] [CrossRef]
- De Galan, B.E.; Perkovic, V.; Ninomiya, T.; Pillai, A.; Patel, A.; Cass, A.; Neal, B.; Poulter, N.; Harrap, S.; Mogensen, C.E.; et al. Lowering blood pressure reduces renal events in type 2 diabetes. J. Am. Soc. Nephrol. 2009, 20, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: Results of the HOPE study and MICRO-HOPE substudy. Lancet 2000, 355, 253–259. [Google Scholar]
- Marre, M.; Lievre, M.; Chatellier, G.; Mann, J.F.; Passa, P.; Menard, J.; Investigators, D.S. Effects of low dose ramipril on cardiovascular and renal outcomes in patients with type 2 diabetes and raised excretion of urinary albumin: Randomised, double blind, placebo controlled trial (the DIABHYCAR study). BMJ 2004, 328, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, C.A.; Fox, K.M.; Remme, W.J.; Bertrand, M.E.; Ferrari, R.; Simoons, M.L.; Investigators, E. The effect of perindopril on cardiovascular morbidity and mortality in patients with diabetes in the EUROPA study: Results from the PERSUADE substudy. Eur. Heart J. 2005, 26, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Thomopoulos, C.; Parati, G.; Zanchetti, A. Effects of blood-pressure-lowering treatment on outcome incidence in hypertension: 10-Should blood pressure management differ in hypertensive patients with and without diabetes mellitus? Overview and meta-analyses of randomized trials. J. Hypertens. 2017, 35, 922–944. [Google Scholar] [CrossRef]
- Catala-Lopez, F.; Macias Saint-Gerons, D.; Gonzalez-Bermejo, D.; Rosano, G.M.; Davis, B.R.; Ridao, M.; Zaragoza, A.; Montero-Corominas, D.; Tobias, A.; de la Fuente-Honrubia, C.; et al. Cardiovascular and Renal Outcomes of Renin-Angiotensin System Blockade in Adult Patients with Diabetes Mellitus: A Systematic Review with Network Meta-Analyses. PLoS Med. 2016, 13, e1001971. [Google Scholar] [CrossRef]
- Vejakama, P.; Thakkinstian, A.; Lertrattananon, D.; Ingsathit, A.; Ngarmukos, C.; Attia, J. Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: A systematic review and network meta-analysis. Diabetologia 2012, 55, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Barnett, A.H.; Bain, S.C.; Bouter, P.; Karlberg, B.; Madsbad, S.; Jervell, J.; Mustonen, J. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N. Engl. J. Med. 2004, 351, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.; Schmieder, R.E.; McQueen, M.; Dyal, L.; Schumacher, H.; Pogue, J.; Wang, X.; Maggioni, A.; Budaj, A.; Chaithiraphan, S.; et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): A multicentre, randomised, double-blind, controlled trial. Lancet 2008, 372, 547–553. [Google Scholar] [CrossRef]
- Wu, H.Y.; Huang, J.W.; Lin, H.J.; Liao, W.C.; Peng, Y.S.; Hung, K.Y.; Wu, K.D.; Tu, Y.K.; Chien, K.L. Comparative effectiveness of renin-angiotensin system blockers and other antihypertensive drugs in patients with diabetes: Systematic review and bayesian network meta-analysis. BMJ 2013, 347, f6008. [Google Scholar] [CrossRef] [Green Version]
- Kunz, R.; Friedrich, C.; Wolbers, M.; Mann, J.F. Meta-analysis: Effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann. Intern. Med. 2008, 148, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, G.F.; Craig, M.; Deeks, J.J.; Schena, F.P.; Craig, J.C. Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: Systematic review. BMJ 2004, 329, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, C.E.; Neldam, S.; Tikkanen, I.; Oren, S.; Viskoper, R.; Watts, R.W.; Cooper, M.E. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: The candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000, 321, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, N.H.; Poulsen, P.L.; Knudsen, S.T.; Poulsen, S.H.; Eiskjaer, H.; Hansen, K.W.; Helleberg, K.; Mogensen, C.E. Long-term dual blockade with candesartan and lisinopril in hypertensive patients with diabetes: The CALM II study. Diabetes Care 2005, 28, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Teo, K.K.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P.; Anderson, C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N. Engl. J. Med. 2008, 358, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F. Supratherapeutic doses of angiotensin receptor blockers to decrease proteinuria in patients with chronic kidney disease. Am. J. Nephrol. 2008, 28, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Persson, F.; Lewis, J.B.; Lewis, E.J.; Hollenberg, N.K. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N. Engl. J. Med. 2008, 358, 2433–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef] [Green Version]
- Chrysostomou, A.; Becker, G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal disease. N. Engl. J. Med. 2001, 345, 925–926. [Google Scholar] [CrossRef]
- Ando, K.; Ohtsu, H.; Uchida, S.; Kaname, S.; Arakawa, Y.; Fujita, T.; Group, E.S. Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: A double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 944–953. [Google Scholar] [CrossRef]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Kolkhof, P.; Borden, S.A. Molecular pharmacology of the mineralocorticoid receptor: Prospects for novel therapeutics. Mol. Cell Endocrinol. 2012, 350, 310–317. [Google Scholar] [CrossRef]
- Sugimoto, K.; Fujimori, A.; Yuyama, H.; Tahara, A.; Fujimura, A. Renal protective effect of YM598, a selective endothelin type A receptor antagonist. J. Cardiovasc. Pharmacol. 2004, 44, S451–S454. [Google Scholar] [CrossRef]
- Feldstein, C.; Romero, C. Role of endothelins in hypertension. Am. J. Ther. 2007, 14, 147–153. [Google Scholar] [CrossRef]
- Granger, J.P.; Abram, S.; Stec, D.; Chandler, D.; LaMarca, B. Endothelin, the kidney, and hypertension. Curr. Hypertens. Rep. 2006, 8, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E. The renal medullary endothelin system in control of sodium and water excretion and systemic blood pressure. Curr. Opin. Nephrol. Hypertens. 2006, 15, 34–40. [Google Scholar] [CrossRef]
- Neuhofer, W.; Pittrow, D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur. J. Clin. Investig. 2006, 36, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.; Kohan, D.E. Physiology and pathology of endothelin-1 in renal mesangium. Am. J. Physiol. Renal Physiol. 2003, 285, F579–F589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.; Zhuo, J.; Alcorn, D.; Casley, D.; Mendelsohn, F.A. Cellular localization of endothelin receptor subtypes in the rat kidney following in vitro labelling. Clin. Exp. Pharmacol. Physiol. 1996, 23, 524–531. [Google Scholar] [CrossRef]
- Cosenzi, A.; Bernobich, E.; Trevisan, R.; Milutinovic, N.; Borri, A.; Bellini, G. Nephroprotective effect of bosentan in diabetic rats. J. Cardiovasc. Pharmacol. 2003, 42, 752–756. [Google Scholar] [CrossRef]
- Kelly, D.J.; Skinner, S.L.; Gilbert, R.E.; Cox, A.J.; Cooper, M.E.; Wilkinson-Berka, J.L. Effects of endothelin or angiotensin II receptor blockade on diabetes in the transgenic (mRen-2)27 rat. Kidney Int. 2000, 57, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.S.; Qiu, C.; Hess, P.; Xi, J.F.; Zheng, N.; Clozel, M. Chronic endothelin receptor blockade prevents both early hyperfiltration and late overt diabetic nephropathy in the rat. J. Cardiovasc. Pharmacol. 2003, 42, 48–54. [Google Scholar] [CrossRef]
- Hocher, B.; Schwarz, A.; Reinbacher, D.; Jacobi, J.; Lun, A.; Priem, F.; Bauer, C.; Neumayer, H.H.; Raschack, M. Effects of endothelin receptor antagonists on the progression of diabetic nephropathy. Nephron 2001, 87, 161–169. [Google Scholar] [CrossRef]
- Sasser, J.M.; Sullivan, J.C.; Hobbs, J.L.; Yamamoto, T.; Pollock, D.M.; Carmines, P.K.; Pollock, J.S. Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J. Am. Soc. Nephrol. 2007, 18, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Pfab, T.; Thone-Reineke, C.; Theilig, F.; Lange, I.; Witt, H.; Maser-Gluth, C.; Bader, M.; Stasch, J.P.; Ruiz, P.; Bachmann, S.; et al. Diabetic endothelin B receptor-deficient rats develop severe hypertension and progressive renal failure. J. Am. Soc. Nephrol. 2006, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, G.M.; Dufresne, J.; Whiteside, C.; Muruve, D.A.; Wong, N.C. Diabetes mellitus increases endothelin-1 gene transcription in rat kidney. Kidney Int. 2000, 58, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Dashwood, M.R.; Mumtaz, F.H.; Thompson, C.S.; Mikhailidis, D.P.; Morgan, R.J. Upregulation of endothelin A receptor sites in the rabbit diabetic kidney: Potential relevance to the early pathogenesis of diabetic nephropathy. Nephron 1999, 83, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Glogowski, E.A.; Tsiani, E.; Zhou, X.; Fantus, I.G.; Whiteside, C. High glucose alters the response of mesangial cell protein kinase C isoforms to endothelin-1. Kidney Int. 1999, 55, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Kohno, M.; Horio, T.; Ikeda, M.; Yokokawa, K.; Fukui, T.; Yasunari, K.; Kurihara, N.; Takeda, T. Angiotensin II stimulates endothelin-1 secretion in cultured rat mesangial cells. Kidney Int. 1992, 42, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Tsiani, E.; Lekas, P.; Fantus, I.G.; Dlugosz, J.; Whiteside, C. High glucose-enhanced activation of mesangial cell p38 MAPK by ET-1, ANG II, and platelet-derived growth factor. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E161–E169. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Goldberg, H.J.; Fantus, I.G.; Whiteside, C.I. High glucose-enhanced mesangial cell extracellular signal-regulated protein kinase activation and alpha1(IV) collagen expression in response to endothelin-1: Role of specific protein kinase C isozymes. Diabetes 2001, 50, 2376–2383. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.K.; Stricklett, P.K.; Padilla, E.; Kohan, D.E. Effect of reactive oxygen species on endothelin-1 production by human mesangial cells. Kidney Int. 1996, 49, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Coll, B.; Andress, D.; Brennan, J.J.; Tang, H.; Houser, M.; Correa-Rotter, R.; Kohan, D.; Lambers Heerspink, H.J.; Makino, H.; et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; Group, A.S. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Langham, R.G.; Kelly, D.J. Urotensin II and the kidney. Curr. Opin. Nephrol. Hypertens. 2013, 22, 107–112. [Google Scholar] [CrossRef]
- Sugo, T.; Murakami, Y.; Shimomura, Y.; Harada, M.; Abe, M.; Ishibashi, Y.; Kitada, C.; Miyajima, N.; Suzuki, N.; Mori, M.; et al. Identification of urotensin II-related peptide as the urotensin II-immunoreactive molecule in the rat brain. Biochem. Biophys. Res. Commun. 2003, 310, 860–868. [Google Scholar] [CrossRef]
- Svistunov, A.A.; Tarasov, V.V.; Shakhmardanova, S.A.; Sologova, S.S.; Bagaturiya, E.T.; Chubarev, V.N.; Galenko-Yaroshevsky, P.A.; Avila-Rodriguez, M.F.; Barreto, G.E.; Aliev, G. Urotensin II: Molecular Mechanisms of Biological Activity. Curr. Protein. Pept. Sci. 2018, 19, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Langham, R.G.; Kelly, D.J.; Gow, R.M.; Zhang, Y.; Dowling, J.K.; Thomson, N.M.; Gilbert, R.E. Increased expression of urotensin II and urotensin II receptor in human diabetic nephropathy. Am. J. Kidney Dis. 2004, 44, 826–831. [Google Scholar] [CrossRef]
- Pang, X.X.; Bai, Q.; Wu, F.; Chen, G.J.; Zhang, A.H.; Tang, C.S. Urotensin II Induces ER Stress and EMT and Increase Extracellular Matrix Production in Renal Tubular Epithelial Cell in Early Diabetic Mice. Kidney Blood Press. Res. 2016, 41, 434–449. [Google Scholar] [CrossRef]
- Chen, G.J.; Wu, F.; Pang, X.X.; Zhang, A.H.; Shi, J.B.; Lu, M.; Tang, C.S. Retraction statement: ‘Urotensin II inhibits autophagy in renal tubular epithelial cells and induces extracellular matrix production in early diabetic mice’ by Guan-Jong Chen, Fei Wu, Xin-Xin Pang, Ai-Hua Zhang, Jun-Bao Shi, Min Lu and Chao-Shu Tang. J. Diabetes Investig. 2017, 8, 629. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Li, C.; Qi, J.; Fu, P.; Yu, X.; Li, X.; Cai, L. Diabetes-induced upregulation of urotensin II and its receptor plays an important role in TGF-beta1-mediated renal fibrosis and dysfunction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1234–E1242. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moyano, M.; Diaz, I.; Dionisio, N.; Zhang, X.; Avila-Medina, J.; Calderon-Sanchez, E.; Trebak, M.; Rosado, J.A.; Ordonez, A.; Smani, T. Urotensin-II promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation. Cardiovasc. Res. 2013, 100, 297–306. [Google Scholar] [CrossRef]
- Song, N.; Ding, W.; Chu, S.; Zhao, J.; Dong, X.; Di, B.; Tang, C. Urotensin II stimulates vascular endothelial growth factor secretion from adventitial fibroblasts in synergy with angiotensin II. Circ. J. 2012, 76, 1267–1273. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, P.; Bousette, N.; Giaid, A. Urotensin-II and cardiovascular remodeling. Peptides 2008, 29, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Totsune, K.; Takahashi, K.; Arihara, Z.; Sone, M.; Murakami, O.; Ito, S.; Kikuya, M.; Ohkubo, T.; Hashimoto, J.; Imai, Y. Elevated plasma levels of immunoreactive urotensin II and its increased urinary excretion in patients with Type 2 diabetes mellitus: Association with progress of diabetic nephropathy. Peptides 2004, 25, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badal, S.S.; Danesh, F.R. New insights into molecular mechanisms of diabetic kidney disease. Am. J. Kidney Dis. 2014, 63, S63–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Ding, D.F.; Chen, S.; Dong, C.L.; Ye, X.L.; Yuan, Y.G.; Feng, Y.M.; You, N.; Xu, J.R.; Miao, H.; et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci. Rep. 2017, 7, 45692. [Google Scholar] [CrossRef]
- Lee, E.Y.; Lee, M.Y.; Hong, S.W.; Chung, C.H.; Hong, S.Y. Blockade of oxidative stress by vitamin C ameliorates albuminuria and renal sclerosis in experimental diabetic rats. Yonsei Med. J. 2007, 48, 847–855. [Google Scholar] [CrossRef]
- Hausse, A.O.; Aggoun, Y.; Bonnet, D.; Sidi, D.; Munnich, A.; Rotig, A.; Rustin, P. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002, 87, 346–349. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic Targeting of the Mitochondria Initiates Excessive Superoxide Production and Mitochondrial Depolarization Causing Decreased mtDNA Integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef] [Green Version]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef] [Green Version]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008, 57, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holterman, C.E.; Thibodeau, J.F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J. Am. Soc. Nephrol. 2014, 25, 784–797. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, S.; Menegazzi, M.; Suzuki, H. Biochemical aspects of nitric oxide. Curr. Pharm. Des. 2004, 10, 1627–1645. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Khaidar, A.; Marx, M.; Lubec, B.; Lubec, G. L-arginine reduces heart collagen accumulation in the diabetic db/db mouse. Circulation 1994, 90, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Komers, R.; Allen, T.J.; Cooper, M.E. Role of endothelium-derived nitric oxide in the pathogenesis of the renal hemodynamic changes of experimental diabetes. Diabetes 1994, 43, 1190–1197. [Google Scholar] [CrossRef]
- Choi, K.C.; Lee, S.C.; Kim, S.W.; Kim, N.H.; Lee, J.U.; Kang, Y.J. Role of nitric oxide in the pathogenesis of diabetic nephropathy in streptozotocin-induced diabetic rats. Korean J. Intern. Med. 1999, 14, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.S. Role of nitric oxide in diabetic nephropathy. Semin. Nephrol. 2004, 24, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Soulis, T.; Cooper, M.E.; Sastra, S.; Thallas, V.; Panagiotopoulos, S.; Bjerrum, O.J.; Jerums, G. Relative contributions of advanced glycation and nitric oxide synthase inhibition to aminoguanidine-mediated renoprotection in diabetic rats. Diabetologia 1997, 40, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, H.; Higuchi, M.; Hora, K. Chronic inhibition of nitric oxide production aggravates diabetic nephropathy in Otsuka Long-Evans Tokushima Fatty rats. Nephron. Physiol. 2006, 104, p12–p22. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolo, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar] [CrossRef] [Green Version]
- Tamae, D.; Lim, P.; Wuenschell, G.E.; Termini, J. Mutagenesis and repair induced by the DNA advanced glycation end product N2-1-(carboxyethyl)-2′-deoxyguanosine in human cells. Biochemistry 2011, 50, 2321–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Randell, E.; Han, Y.; Adeli, K.; Krahn, J.; Meng, Q.H. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin. Biochem. 2011, 44, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nakayama, M.; Iwabuchi, M.; Terawaki, H.; Sato, T.; Kohno, M.; Ito, S. Plasma alpha-oxoaldehyde levels in diabetic and nondiabetic chronic kidney disease patients. Am. J. Nephrol. 2008, 28, 871–878. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Drummond, K.S.; Nelson, R.G.; Howell, S.K.; Szwergold, B.S.; Mauer, M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 2005, 54, 3274–3281. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.J.; Scheijen, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease in Individuals With Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66, 2278–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanssen, N.M.J.; Westerink, J.; Scheijen, J.; van der Graaf, Y.; Stehouwer, C.D.A.; Schalkwijk, C.G.; Group, S.S. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease and Mortality in Individuals With Type 2 Diabetes. Diabetes Care 2018, 41, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.M.; Vistisen, D.; Fleming, T.; Nawroth, P.P.; Rossing, P.; Jorgensen, M.E.; Lauritzen, T.; Sandbaek, A.; Witte, D.R. Methylglyoxal is associated with changes in kidney function among individuals with screen-detected Type 2 diabetes mellitus. Diabet. Med. 2016, 33, 1625–1631. [Google Scholar] [CrossRef]
- Rabbani, N.; Sebekova, K.; Sebekova, K., Jr.; Heidland, A.; Thornalley, P.J. Accumulation of free adduct glycation, oxidation, and nitration products follows acute loss of renal function. Kidney Int. 2007, 72, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Kim, Y.S.; Jung, D.H.; Lee, J.; Kim, J.S. Increased glyoxalase I levels inhibit accumulation of oxidative stress and an advanced glycation end product in mouse mesangial cells cultured in high glucose. Exp. Cell Res. 2012, 318, 152–159. [Google Scholar] [CrossRef]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Ostergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
- Kusunoki, H.; Miyata, S.; Ohara, T.; Liu, B.F.; Uriuhara, A.; Kojima, H.; Suzuki, K.; Miyazaki, H.; Yamashita, Y.; Inaba, K.; et al. Relation between serum 3-deoxyglucosone and development of diabetic microangiopathy. Diabetes Care 2003, 26, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; Group, D.E.R.; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, R.; Bohme, D.; Singer, D.; Frolov, A. Specific tandem mass spectrometric detection of AGE-modified arginine residues in peptides. J. Mass Spectrom. 2015, 50, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Soulis, T.; Cooper, M.E.; Vranes, D.; Bucala, R.; Jerums, G. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney Int. 1996, 50, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulis-Liparota, T.; Cooper, M.; Papazoglou, D.; Clarke, B.; Jerums, G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion, and tissue fluorescence in streptozocin-induced diabetic rat. Diabetes 1991, 40, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Martin, S.; Federlin, K.; Geisen, K.; Brownlee, M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 1991, 88, 11555–11558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, M.; Schmelzer, J.D.; Poduslo, J.F.; Curran, G.L.; Nickander, K.K.; Low, P.A. Aminoguanidine effects on nerve blood flow, vascular permeability, electrophysiology, and oxygen free radicals. Proc. Natl. Acad. Sci. USA 1991, 88, 6107–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Wuerth, J.P.; Cartwright, K.; Bain, R.P.; Dippe, S.; Hershon, K.; Mooradian, A.D.; Spinowitz, B.S. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control. Clin. Trials 1999, 20, 493–510. [Google Scholar] [CrossRef]
- Lobner, J.; Degen, J.; Henle, T. Creatine is a scavenger for methylglyoxal under physiological conditions via formation of N-(4-methyl-5-oxo-1-imidazolin-2-yl)sarcosine (MG-HCr). J. Agric. Food Chem. 2015, 63, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Stehouwer, C.D.; Schalkwijk, C.G. Current therapeutic interventions in the glycation pathway: Evidence from clinical studies. Diabetes Obes. Metab. 2013, 15, 677–689. [Google Scholar] [CrossRef]
- Little, W.C.; Zile, M.R.; Kitzman, D.W.; Hundley, W.G.; O’Brien, T.X.; Degroof, R.C. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J. Card. Fail. 2005, 11, 191–195. [Google Scholar] [CrossRef]
- Zieman, S.J.; Melenovsky, V.; Clattenburg, L.; Corretti, M.C.; Capriotti, A.; Gerstenblith, G.; Kass, D.A. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J. Hypertens. 2007, 25, 577–583. [Google Scholar] [CrossRef]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine as a multifunctional pharmaceutical: Targeting pathogenic glycation and oxidative damage. Cell Mol. Life Sci. 2005, 62, 1671–1681. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Lewis, E.J.; Greene, T.; Spitalewiz, S.; Blumenthal, S.; Berl, T.; Hunsicker, L.G.; Pohl, M.A.; Rohde, R.D.; Raz, I.; Yerushalmy, Y.; et al. Pyridorin in type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Giannarelli, R.; Aragona, M.; Coppelli, A.; Del Prato, S. Reducing insulin resistance with metformin: The evidence today. Diabetes Metab. 2003, 29, 6S28–6S35. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. (Lond.) 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero-Lopez, D.; Lecomte, M.; Moinet, G.; Patereau, G.; Lagarde, M.; Wiernsperger, N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem. Pharmacol. 1999, 58, 1765–1773. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Kender, Z.; Fleming, T.; Kopf, S.; Torzsa, P.; Grolmusz, V.; Herzig, S.; Schleicher, E.; Racz, K.; Reismann, P.; Nawroth, P.P. Effect of metformin on methylglyoxal metabolism in patients with type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2014, 122, 316–319. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal To Form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.; Jensen, J.B.; Jakobsen, S.; Jessen, N.; Frokiaer, J.; Kemp, B.E.; Marciszyn, A.L.; Li, H.; Pastor-Soler, N.M.; Hallows, K.R.; et al. Renoprotective Effects of Metformin are Independent of Organic Cation Transporters 1 & 2 and AMP-activated Protein Kinase in the Kidney. Sci. Rep. 2016, 6, 35952. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.S.; Cheng, Y.H.; Chiou, C.H.; Chang, T.L. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J. Agric. Food Chem. 2012, 60, 9180–9187. [Google Scholar] [CrossRef]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef]
- Yeh, W.J.; Hsia, S.M.; Lee, W.H.; Wu, C.H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef]
- Van den Eynde, M.D.G.; Geleijnse, J.M.; Scheijen, J.; Hanssen, N.M.J.; Dower, J.I.; Afman, L.A.; Stehouwer, C.D.A.; Hollman, P.C.H.; Schalkwijk, C.G. Quercetin, but Not Epicatechin, Decreases Plasma Concentrations of Methylglyoxal in Adults in a Randomized, Double-Blind, Placebo-Controlled, Crossover Trial with Pure Flavonoids. J. Nutr. 2018, 148, 1911–1916. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, T.; Sang, S.; Lv, L. Quercetin inhibits advanced glycation end product formation by trapping methylglyoxal and glyoxal. J. Agric. Food Chem. 2014, 62, 12152–12158. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, B.N.; Kim, Y.W.; Keum, Y.S. Mechanisms of Nrf2/Keap1-dependent phase II cytoprotective and detoxifying gene expression and potential cellular targets of chemopreventive isothiocyanates. Oxid. Med. Cell. Longev. 2013, 2013, 839409. [Google Scholar] [CrossRef] [Green Version]
- Mann, G.E.; Niehueser-Saran, J.; Watson, A.; Gao, L.; Ishii, T.; de Winter, P.; Siow, R.C. Nrf2/ARE regulated antioxidant gene expression in endothelial and smooth muscle cells in oxidative stress: Implications for atherosclerosis and preeclampsia. Sheng Li Xue Bao 2007, 59, 117–127. [Google Scholar]
- Hsu, W.H.; Lee, B.H.; Chang, Y.Y.; Hsu, Y.W.; Pan, T.M. A novel natural Nrf2 activator with PPARgamma-agonist (monascin) attenuates the toxicity of methylglyoxal and hyperglycemia. Toxicol. Appl. Pharmacol. 2013, 272, 842–851. [Google Scholar] [CrossRef]
- Alfarano, M.; Pastore, D.; Fogliano, V.; Schalkwijk, C.G.; Oliviero, T. The Effect of Sulforaphane on Glyoxalase I Expression and Activity in Peripheral Blood Mononuclear Cells. Nutrients 2018, 10, 1773. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Glyoxalase 1 Modulation in Obesity and Diabetes. Antioxid. Redox Signal. 2019, 30, 354–374. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, M.T.; Yap, F.Y.; Tong, D.C.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Cassese, A.; Esposito, I.; Fiory, F.; Barbagallo, A.P.; Paturzo, F.; Mirra, P.; Ulianich, L.; Giacco, F.; Iadicicco, C.; Lombardi, A.; et al. In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J. Biol. Chem. 2008, 283, 36088–36099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.A.; Li, S.L.; Sahar, S.; Kim, Y.S.; Xu, Z.G.; Lanting, L.; Natarajan, R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J. Biol. Chem. 2006, 281, 13685–13693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalfa, T.A.; Gerritsen, M.E.; Carlson, E.C.; Binstock, A.J.; Tsilibary, E.C. Altered proliferation of retinal microvascular cells on glycated matrix. Investig. Ophthalmol. Vis. Sci. 1995, 36, 2358–2367. [Google Scholar]
- Mott, J.D.; Khalifah, R.G.; Nagase, H.; Shield, C.F., 3rd; Hudson, J.K.; Hudson, B.G. Nonenzymatic glycation of type IV collagen and matrix metalloproteinase susceptibility. Kidney Int. 1997, 52, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-beta and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.X.; Wells-Knecht, K.J.; Blackledge, J.A.; Lyons, T.J.; Thorpe, S.R.; Baynes, J.W. Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes 1994, 43, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Liang, J.T.; Tsai, P.S.; Wu, M.S.; Hsu, K.L. Prevention of arterial stiffening by pyridoxamine in diabetes is associated with inhibition of the pathogenic glycation on aortic collagen. Br. J. Pharmacol. 2009, 157, 1419–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, B.; Duriez, M.; Poitevin, P.; Heudes, D.; Bruneval, P.; Tedgui, A.; Levy, B.I. Aminoguanidine prevents age-related arterial stiffening and cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 1998, 95, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Kass, D.A.; Shapiro, E.P.; Kawaguchi, M.; Capriotti, A.R.; Scuteri, A.; deGroof, R.C.; Lakatta, E.G. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation 2001, 104, 1464–1470. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sourris, K.C.; Forbes, J.M. Interactions between advanced glycation end-products (AGE) and their receptors in the development and progression of diabetic nephropathy—Are these receptors valid therapeutic targets. Curr. Drug Targets 2009, 10, 42–50. [Google Scholar] [CrossRef]
- Sundblad, V.; Croci, D.O.; Rabinovich, G.A. Regulated expression of galectin-3, a multifunctional glycan-binding protein, in haematopoietic and non-haematopoietic tissues. Histol. Histopathol. 2011, 26, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Yan, S.D.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira-Machado, J.A.; Volpe, C.M.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: A functional tripod that leads to diabetic inflammation. Expert. Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef]
- Heizmann, C.W.; Ackermann, G.E.; Galichet, A. Pathologies involving the S100 proteins and RAGE. Subcell. Biochem. 2007, 45, 93–138. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol 2003, 162, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Isoda, K.; Folco, E.; Marwali, M.R.; Ohsuzu, F.; Libby, P. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis 2008, 198, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.; Inagaki, Y.; Okamoto, T.; Amano, S.; Koga, K.; Takeuchi, M.; Makita, Z. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J. Biol. Chem. 2002, 277, 20309–20315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef]
- Tsuchida, K.; Makita, Z.; Yamagishi, S.; Atsumi, T.; Miyoshi, H.; Obara, S.; Ishida, M.; Ishikawa, S.; Yasumura, K.; Koike, T. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia 1999, 42, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Inagaki, Y.; Okamoto, T.; Amano, S.; Koga, K.; Takeuchi, M. Advanced glycation end products inhibit de novo protein synthesis and induce TGF-beta overexpression in proximal tubular cells. Kidney Int. 2003, 63, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008, 283, 35507–35516. [Google Scholar] [CrossRef] [Green Version]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S.; Charlton-Menys, V.; Bao, W.; Demicco, D.A.; Preston, G.M.; et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: An analysis from the CARDS trial. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpert, P.M.; Djuric, Z.; Kopf, S.; Rudofsky, G.; Morcos, M.; Nawroth, P.P.; Bierhaus, A. Soluble RAGE but not endogenous secretory RAGE is associated with albuminuria in patients with type 2 diabetes. Cardiovasc. Diabetol. 2007, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nin, J.W.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Chaturvedi, N.; Fuller, J.H.; Stehouwer, C.D.; Group, E.P.C.S. Levels of soluble receptor for AGE are cross-sectionally associated with cardiovascular disease in type 1 diabetes, and this association is partially mediated by endothelial and renal dysfunction and by low-grade inflammation: The EURODIAB Prospective Complications Study. Diabetologia 2009, 52, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.C.; Soderlund, J.; Lehto, M.; Makinen, V.P.; Moran, J.L.; Cooper, M.E.; Forsblom, C.; Groop, P.H.; FinnDiane Study Group. Soluble receptor for AGE (RAGE) is a novel independent predictor of all-cause and cardiovascular mortality in type 1 diabetes. Diabetologia 2011, 54, 2669–2677. [Google Scholar] [CrossRef]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher plasma soluble Receptor for Advanced Glycation End Products (sRAGE) levels are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes 2010, 59, 2027–2032. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.K.; Keenan, H.A.; Cavallerano, J.D.; Asztalos, B.F.; Schaefer, E.J.; Sell, D.R.; Strauch, C.M.; Monnier, V.M.; Doria, A.; Aiello, L.P.; et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: The joslin 50-year medalist study. Diabetes Care 2011, 34, 968–974. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.X.; Lu, L.; Peng, W.H.; Wang, L.J.; Zhang, Q.; Zhang, R.Y.; Chen, Q.J.; Shen, W.F. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 2009, 205, 544–548. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Patel, S.K.; Jerums, G.; Penfold, S.A.; Nguyen, T.V.; Sourris, K.C.; Panagiotopoulos, S.; Srivastava, P.M.; Cooper, M.E.; Burrell, L.M.; et al. Advanced glycation urinary protein-bound biomarkers and severity of diabetic nephropathy in man. Am. J. Nephrol. 2011, 34, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Friess, U.; Waldner, M.; Wahl, H.G.; Lehmann, R.; Haring, H.U.; Voelter, W.; Schleicher, E. Liquid chromatography-based determination of urinary free and total N(epsilon)-(carboxymethyl)lysine excretion in normal and diabetic subjects. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 794, 273–280. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; van Ypersele de Strihou, C.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Francis, J. Pyridoxamine, advanced glycation inhibition, and diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Alam, S.S.; Riaz, S.; Larkin, J.R.; Akhtar, M.W.; Shafi, T.; Thornalley, P.J. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: A randomised, double-blind placebo-controlled pilot study. Diabetologia 2009, 52, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppa, M.; Brem, H.; Cai, W.; Zhang, J.G.; Basgen, J.; Li, Z.; Vlassara, H.; Uribarri, J. Prevention and reversal of diabetic nephropathy in db/db mice treated with alagebrium (ALT-711). Am. J. Nephrol. 2006, 26, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Makita, Z.; Ishikawa, S.; Yasumura, K.; Fujii, W.; Yanagisawa, K.; Kawata, T.; Koike, T. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes 1997, 46, 895–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef]
- Galasko, D.; Bell, J.; Mancuso, J.Y.; Kupiec, J.W.; Sabbagh, M.N.; van Dyck, C.; Thomas, R.G.; Aisen, P.S.; Alzheimer’s Disease Cooperative, S. Clinical trial of an inhibitor of RAGE-Abeta interactions in Alzheimer disease. Neurology 2014, 82, 1536–1542. [Google Scholar] [CrossRef]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Myint, K.M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K.; et al. RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [Green Version]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.L.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, A.A.; Capobianco, A.; Esposito, A.; De Cobelli, F.; Canu, T.; Monno, A.; Raucci, A.; Sanvito, F.; Doglioni, C.; Nawroth, P.P.; et al. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J. Immunol. 2008, 180, 2270–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, B.; Desai, D.D.; Downie, M.P.; Chen, Y.; Yan, S.F.; Herold, K.; Schmidt, A.M.; Clynes, R. Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J. Immunol. 2007, 179, 8051–8058. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorpe, S.R.; Thallas-Bonke, V.; Pete, J.; Thomas, M.C.; Deemer, E.R.; Bassal, S.; El-Osta, A.; Long, D.M.; Panagiotopoulos, S.; et al. Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2363–2372. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Ueda, S.; Yamagishi, S.; Kato, S.; Inagaki, Y.; Takeuchi, M.; Motomiya, Y.; Bucala, R.; Iida, S.; Tamaki, K.; et al. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004, 66, 2137–2147. [Google Scholar] [CrossRef] [Green Version]
- Pickering, R.J.; Tikellis, C.; Rosado, C.J.; Tsorotes, D.; Dimitropoulos, A.; Smith, M.; Huet, O.; Seeber, R.M.; Abhayawardana, R.; Johnstone, E.K.; et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J. Clin. Investig. 2019, 129, 406–421. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A. Sodium-glucose cotransporter 2 inhibition in type 1 diabetes: Simultaneous glucose lowering and renal protection? Can. J. Diabetes 2014, 38, 356–363. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Perkins, B.A.; Cherney, D.Z.; Partridge, H.; Soleymanlou, N.; Tschirhart, H.; Zinman, B.; Fagan, N.M.; Kaspers, S.; Woerle, H.J.; Broedl, U.C.; et al. Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: Results of an 8-week open-label proof-of-concept trial. Diabetes Care 2014, 37, 1480–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, R.C. Sodium glucose transport 2 (SGLT2) inhibition decreases glomerular hyperfiltration: Is there a role for SGLT2 inhibitors in diabetic kidney disease? Circulation 2014, 129, 542–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.J.; Lee, T.; DeFronzo, R.A. Why Do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in humans? Diabetes 2012, 61, 2199–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol. Renal Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef] [Green Version]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef]
- Yale, J.F.; Bakris, G.; Cariou, B.; Yue, D.; David-Neto, E.; Xi, L.; Figueroa, K.; Wajs, E.; Usiskin, K.; Meininger, G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2013, 15, 463–473. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2099. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef] [PubMed]
- Rajasekeran, H.; Lytvyn, Y.; Bozovic, A.; Lovshin, J.A.; Diamandis, E.; Cattran, D.; Husain, M.; Perkins, B.A.; Advani, A.; Reich, H.N.; et al. Urinary adenosine excretion in type 1 diabetes. Am. J. Physiol. Renal Physiol. 2017, 313, F184–F191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.W.; Kim, H.W.; Ko, S.H.; Lim, J.H.; Ryu, G.R.; Chung, H.W.; Han, S.W.; Shin, S.J.; Bang, B.K.; Breyer, M.D.; et al. Long-term treatment of glucagon-like peptide-1 analog exendin-4 ameliorates diabetic nephropathy through improving metabolic anomalies in db/db mice. J. Am. Soc. Nephrol. 2007, 18, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Xie, S.H.; Liu, Y.N.; Kim, W.; Jin, H.Y.; Park, S.K.; Shao, Y.M.; Park, T.S. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J. Pharmacol. Exp. Ther. 2012, 340, 248–255. [Google Scholar] [CrossRef]
- Thomas, M.C. The potential and pitfalls of GLP-1 receptor agonists for renal protection in type 2 diabetes. Diabetes Metab. 2017, 43, 2S20–2S27. [Google Scholar] [CrossRef]
- Gutzwiller, J.P.; Tschopp, S.; Bock, A.; Zehnder, C.E.; Huber, A.R.; Kreyenbuehl, M.; Gutmann, H.; Drewe, J.; Henzen, C.; Goeke, B.; et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J. Clin. Endocrinol. Metab. 2004, 89, 3055–3061. [Google Scholar] [CrossRef]
- Tonneijck, L.; Smits, M.M.; Muskiet, M.H.A.; Hoekstra, T.; Kramer, M.H.H.; Danser, A.H.J.; Diamant, M.; Joles, J.A.; van Raalte, D.H. Acute renal effects of the GLP-1 receptor agonist exenatide in overweight type 2 diabetes patients: A randomised, double-blind, placebo-controlled trial. Diabetologia 2016, 59, 1412–1421. [Google Scholar] [CrossRef] [Green Version]
- Skov, J.; Dejgaard, A.; Frokiaer, J.; Holst, J.J.; Jonassen, T.; Rittig, S.; Christiansen, J.S. Glucagon-like peptide-1 (GLP-1): Effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J. Clin. Endocrinol. Metab. 2013, 98, E664–E671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, J.; Pedersen, M.; Holst, J.J.; Madsen, B.; Goetze, J.P.; Rittig, S.; Jonassen, T.; Frokiaer, J.; Dejgaard, A.; Christiansen, J.S. Short-term effects of liraglutide on kidney function and vasoactive hormones in type 2 diabetes: A randomized clinical trial. Diabetes Obes. Metab. 2016, 18, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Moreno, C.; Hoagland, K.M.; Dahly, A.; Ditter, K.; Mistry, M.; Roman, R.J. Antihypertensive effect of glucagon-like peptide 1 in Dahl salt-sensitive rats. J. Hypertens. 2003, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, M.H.A.; Tonneijck, L.; Smits, M.M.; van Baar, M.J.B.; Kramer, M.H.H.; Hoorn, E.J.; Joles, J.A.; van Raalte, D.H. GLP-1 and the kidney: From physiology to pharmacology and outcomes in diabetes. Nat. Rev. Nephrol. 2017, 13, 605–628. [Google Scholar] [CrossRef]
- Davies, M.J.; Bergenstal, R.; Bode, B.; Kushner, R.F.; Lewin, A.; Skjoth, T.V.; Andreasen, A.H.; Jensen, C.B.; DeFronzo, R.A.; Group, N.N.S. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. JAMA 2015, 314, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.E.; Orsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornoe, K.; Zinman, B.; Buse, J.B.; Committee, L.S.; et al. Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Muskiet, M.H.A.; Tonneijck, L.; Huang, Y.; Liu, M.; Saremi, A.; Heerspink, H.J.L.; van Raalte, D.H. Lixisenatide and renal outcomes in patients with type 2 diabetes and acute coronary syndrome: An exploratory analysis of the ELIXA randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2018, 6, 859–869. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Hu, C.; Lu, W. Exenatide reduces urinary transforming growth factor-beta1 and type IV collagen excretion in patients with type 2 diabetes and microalbuminuria. Kidney Blood Press. Res. 2012, 35, 483–488. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Tuttle, K.R.; McKinney, T.D.; Davidson, J.A.; Anglin, G.; Harper, K.D.; Botros, F.T. Effects of once-weekly dulaglutide on kidney function in patients with type 2 diabetes in phase II and III clinical trials. Diabetes Obes. Metab. 2017, 19, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, K.R.; Lakshmanan, M.C.; Rayner, B.; Busch, R.S.; Zimmermann, A.G.; Woodward, D.B.; Botros, F.T. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): A multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 605–617. [Google Scholar] [CrossRef]
- Lovshin, J.A.; Rajasekeran, H.; Lytvyn, Y.; Lovblom, L.E.; Khan, S.; Alemu, R.; Locke, A.; Lai, V.; He, H.; Hittle, L.; et al. Dipeptidyl Peptidase 4 Inhibition Stimulates Distal Tubular Natriuresis and Increases in Circulating SDF-1alpha(1-67) in Patients With Type 2 Diabetes. Diabetes Care 2017, 40, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonneijck, L.; Smits, M.M.; Muskiet, M.H.; Hoekstra, T.; Kramer, M.H.; Danser, A.H.; Ter Wee, P.M.; Diamant, M.; Joles, J.A.; van Raalte, D.H. Renal Effects of DPP-4 Inhibitor Sitagliptin or GLP-1 Receptor Agonist Liraglutide in Overweight Patients With Type 2 Diabetes: A 12-Week, Randomized, Double-Blind, Placebo-Controlled Trial. Diabetes Care 2016, 39, 2042–2050. [Google Scholar] [CrossRef] [Green Version]
- Takashima, S.; Fujita, H.; Fujishima, H.; Shimizu, T.; Sato, T.; Morii, T.; Tsukiyama, K.; Narita, T.; Takahashi, T.; Drucker, D.J.; et al. Stromal cell-derived factor-1 is upregulated by dipeptidyl peptidase-4 inhibition and has protective roles in progressive diabetic nephropathy. Kidney Int. 2016, 90, 783–796. [Google Scholar] [CrossRef]
- Alter, M.L.; Ott, I.M.; von Websky, K.; Tsuprykov, O.; Sharkovska, Y.; Krause-Relle, K.; Raila, J.; Henze, A.; Klein, T.; Hocher, B. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press. Res. 2012, 36, 119–130. [Google Scholar] [CrossRef]
- Gangadharan Komala, M.; Gross, S.; Zaky, A.; Pollock, C.; Panchapakesan, U. Saxagliptin reduces renal tubulointerstitial inflammation, hypertrophy and fibrosis in diabetes. Nephrology (Carlton) 2016, 21, 423–431. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Groop, P.H.; Cooper, M.E.; Perkovic, V.; Emser, A.; Woerle, H.J.; von Eynatten, M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care 2013, 36, 3460–3468. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.E.; Perkovic, V.; McGill, J.B.; Groop, P.H.; Wanner, C.; Rosenstock, J.; Hehnke, U.; Woerle, H.J.; von Eynatten, M. Kidney Disease End Points in a Pooled Analysis of Individual Patient-Level Data From a Large Clinical Trials Program of the Dipeptidyl Peptidase 4 Inhibitor Linagliptin in Type 2 Diabetes. Am. J. Kidney Dis. 2015, 66, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Groop, P.H.; Cooper, M.E.; Perkovic, V.; Hocher, B.; Kanasaki, K.; Haneda, M.; Schernthaner, G.; Sharma, K.; Stanton, R.C.; Toto, R.; et al. Linagliptin and its effects on hyperglycaemia and albuminuria in patients with type 2 diabetes and renal dysfunction: The randomized MARLINA-T2D trial. Diabetes Obes. Metab. 2017, 19, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Cornel, J.H.; Bakris, G.L.; Stevens, S.R.; Alvarsson, M.; Bax, W.A.; Chuang, L.M.; Engel, S.S.; Lopes, R.D.; McGuire, D.K.; Riefflin, A.; et al. Effect of Sitagliptin on Kidney Function and Respective Cardiovascular Outcomes in Type 2 Diabetes: Outcomes From TECOS. Diabetes Care 2016, 39, 2304–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosenzon, O.; Leibowitz, G.; Bhatt, D.L.; Cahn, A.; Hirshberg, B.; Wei, C.; Im, K.; Rozenberg, A.; Yanuv, I.; Stahre, C.; et al. Effect of Saxagliptin on Renal Outcomes in the SAVOR-TIMI 53 Trial. Diabetes Care 2017, 40, 69–76. [Google Scholar] [CrossRef] [Green Version]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Vlagopoulos, P.T.; Tighiouart, H.; Weiner, D.E.; Griffith, J.; Pettitt, D.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. Anemia as a risk factor for cardiovascular disease and all-cause mortality in diabetes: The impact of chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 3403–3410. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Hasegawa, G.; Tanaka, M.; Osaka, T.; Shiotsu, Y.; Narumiya, H.; Inoue, M.; Nakano, K.; Nakamura, N.; Fukui, M. Association between Hemoglobin Concentration and the Progression or Development of Albuminuria in Diabetic Kidney Disease. PLoS ONE 2015, 10, e0129192. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, M.A.; Burdmann, E.A.; Chen, C.Y.; Cooper, M.E.; de Zeeuw, D.; Eckardt, K.U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N. Engl. J. Med. 2009, 361, 2019–2032. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
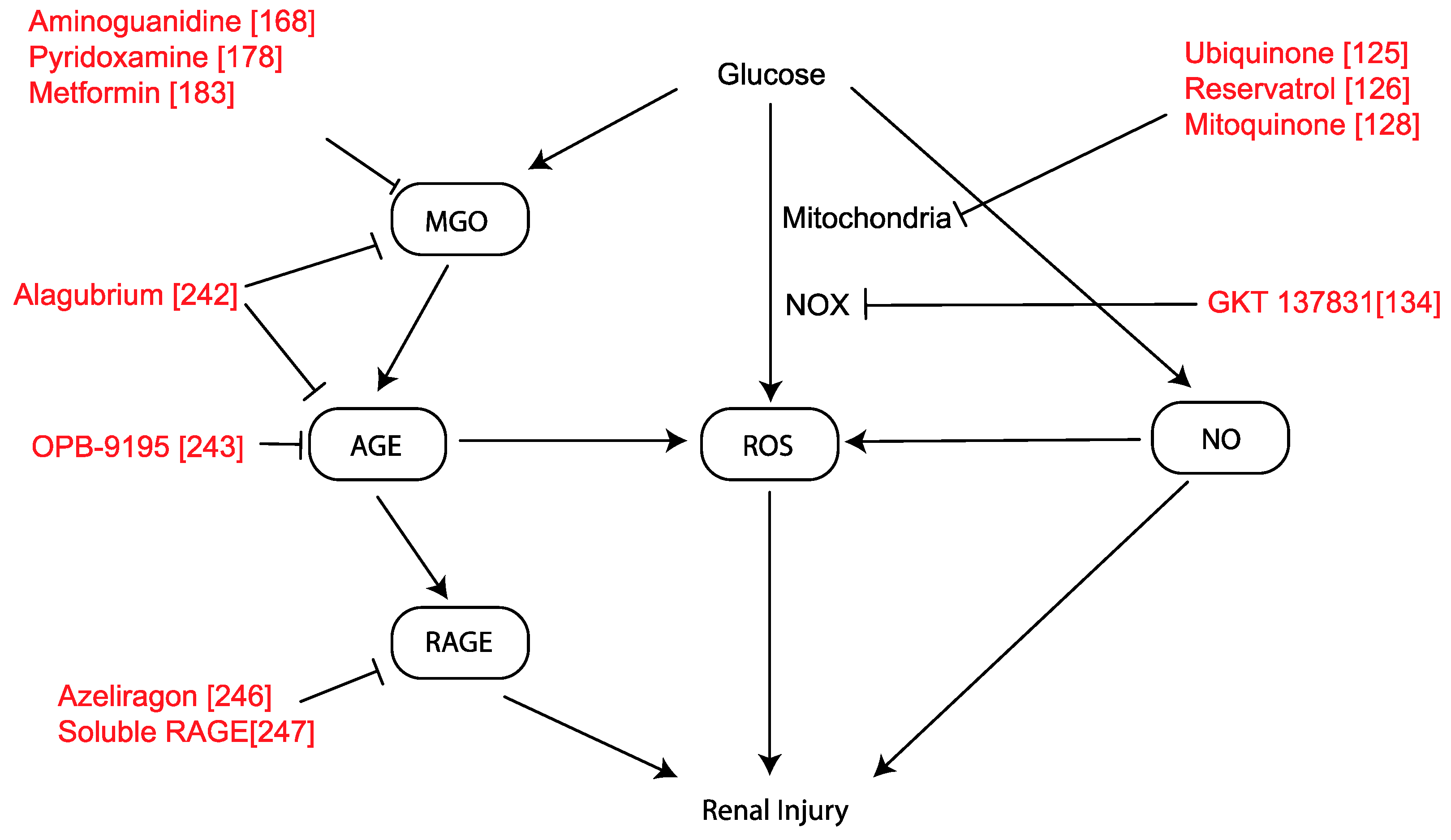{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.M.; Bose, M.; Cooper, M.E. Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 2218. https://doi.org/10.3390/ijms21062218
Patel DM, Bose M, Cooper ME. Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease. International Journal of Molecular Sciences. 2020; 21(6):2218. https://doi.org/10.3390/ijms21062218
Chicago/Turabian StylePatel, Devang M., Madhura Bose, and Mark E. Cooper. 2020. "Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease" International Journal of Molecular Sciences 21, no. 6: 2218. https://doi.org/10.3390/ijms21062218
APA StylePatel, D. M., Bose, M., & Cooper, M. E. (2020). Glucose and Blood Pressure-Dependent Pathways–The Progression of Diabetic Kidney Disease. International Journal of Molecular Sciences, 21(6), 2218. https://doi.org/10.3390/ijms21062218