Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of RNA-Binding Proteins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
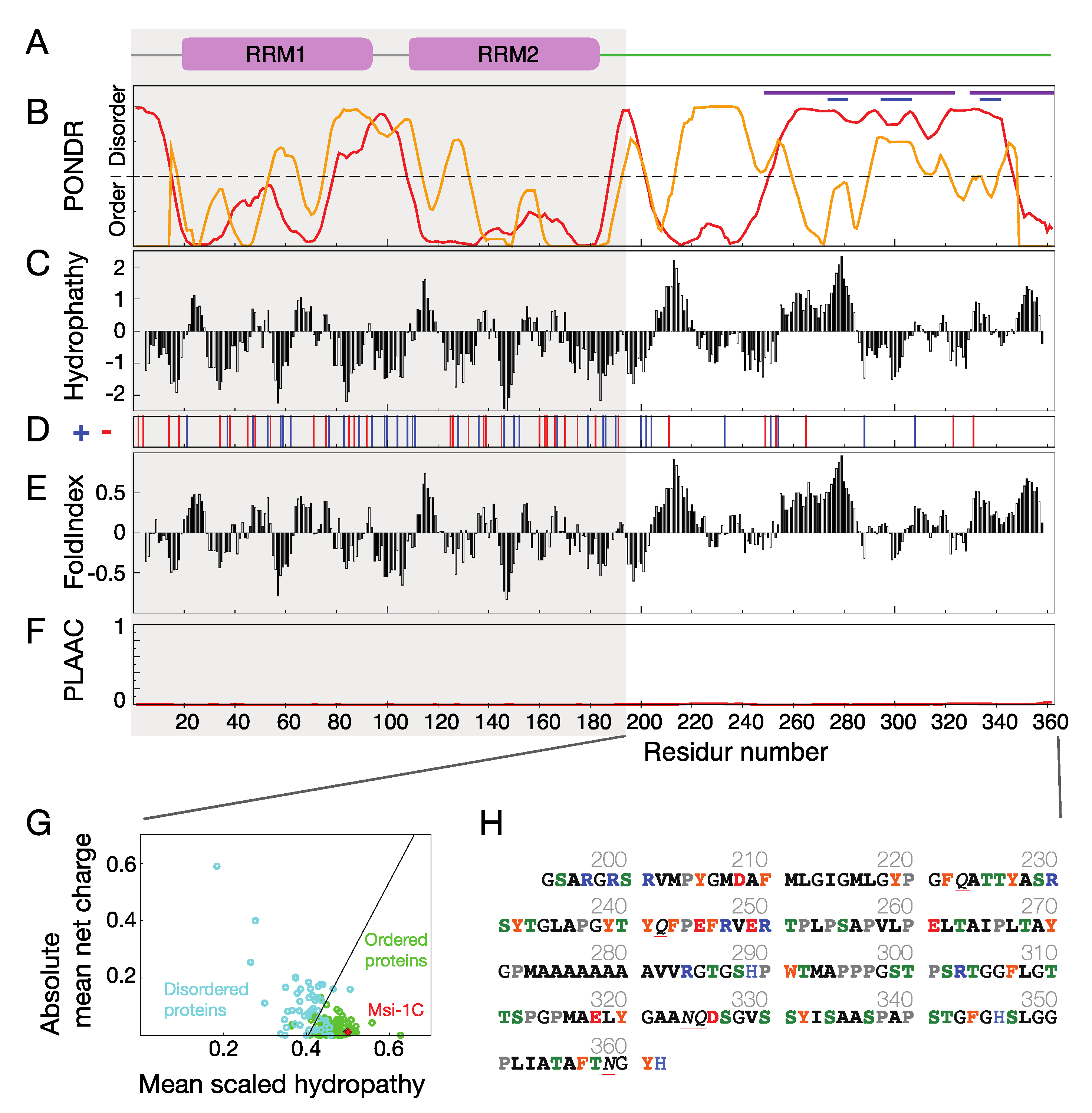
:1. Introduction
2. Results and Discussion
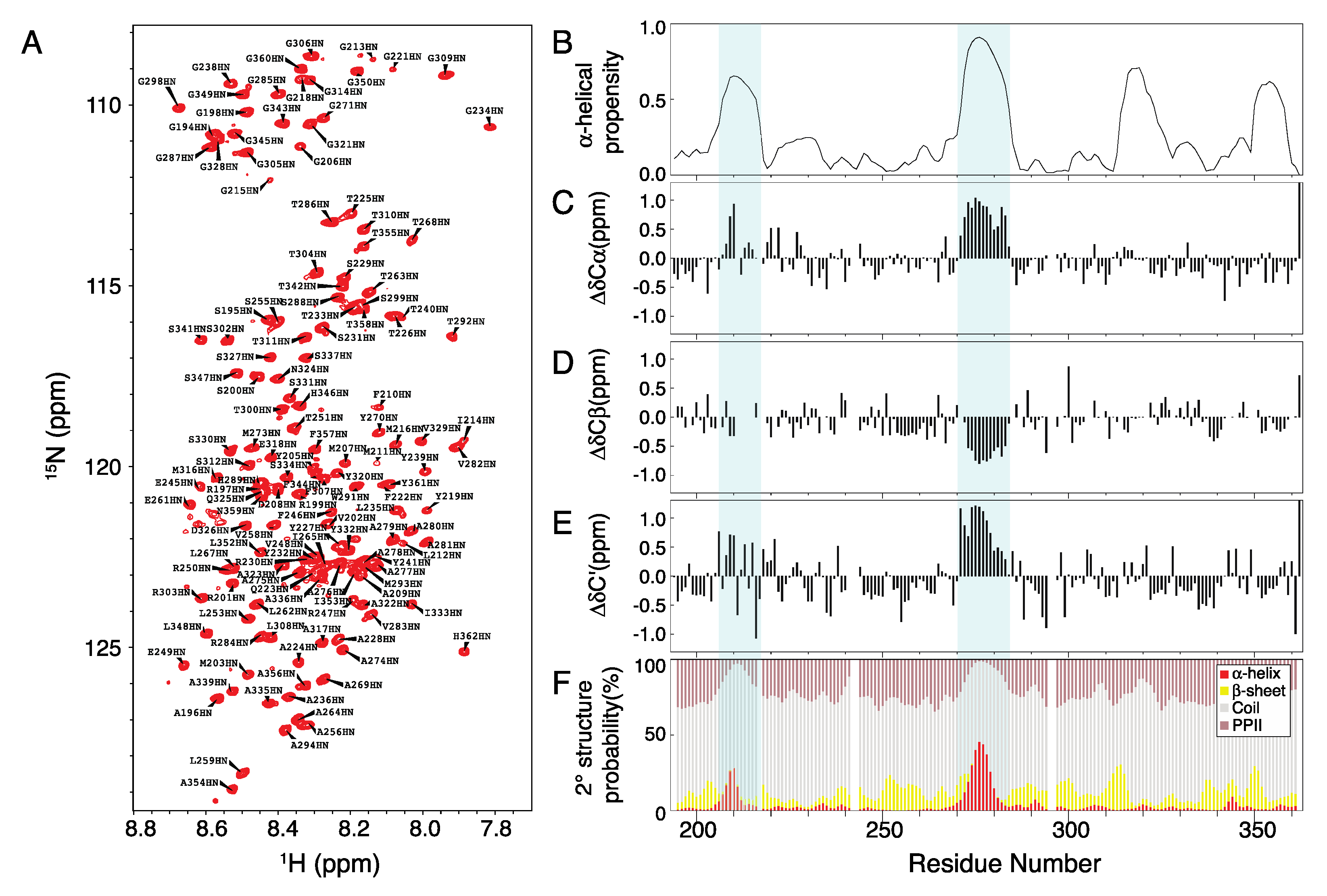
2.1. Chemical Shift Assignment of Musashi-1
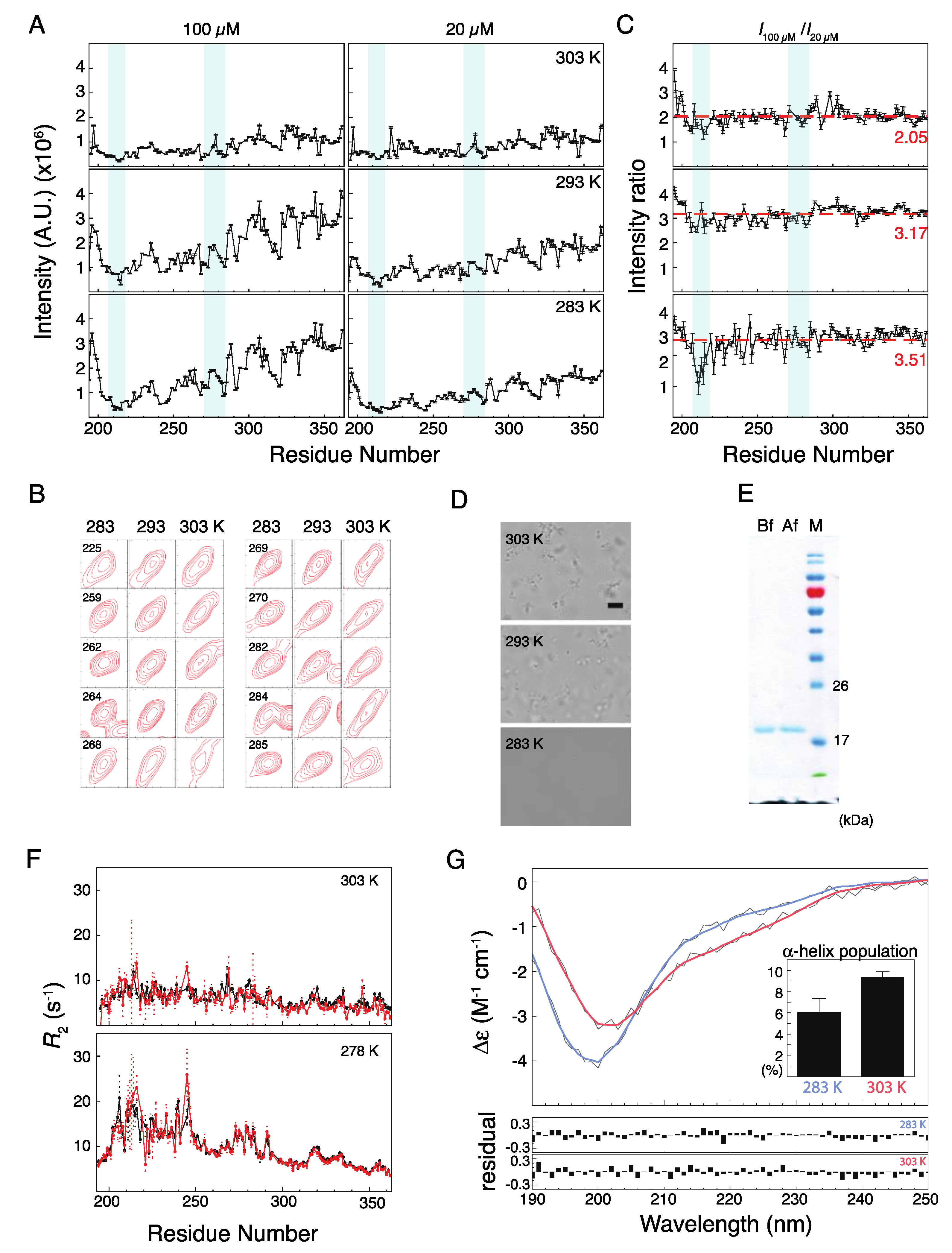
2.2. Musashi-1 Self-Association
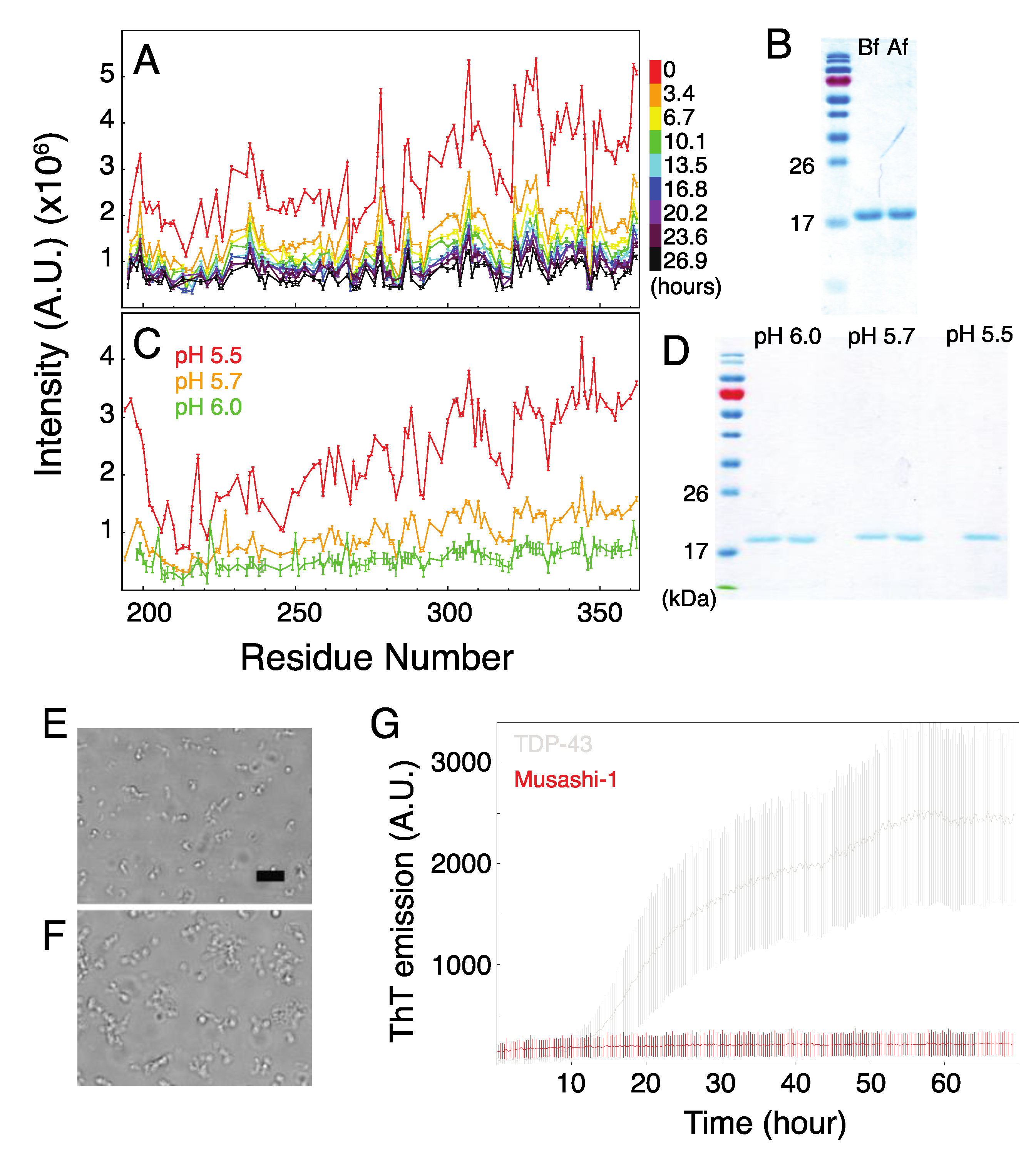
2.3. Formation of Musashi-1 Oligomers in a Time-Dependent Manner and at a Higher pH
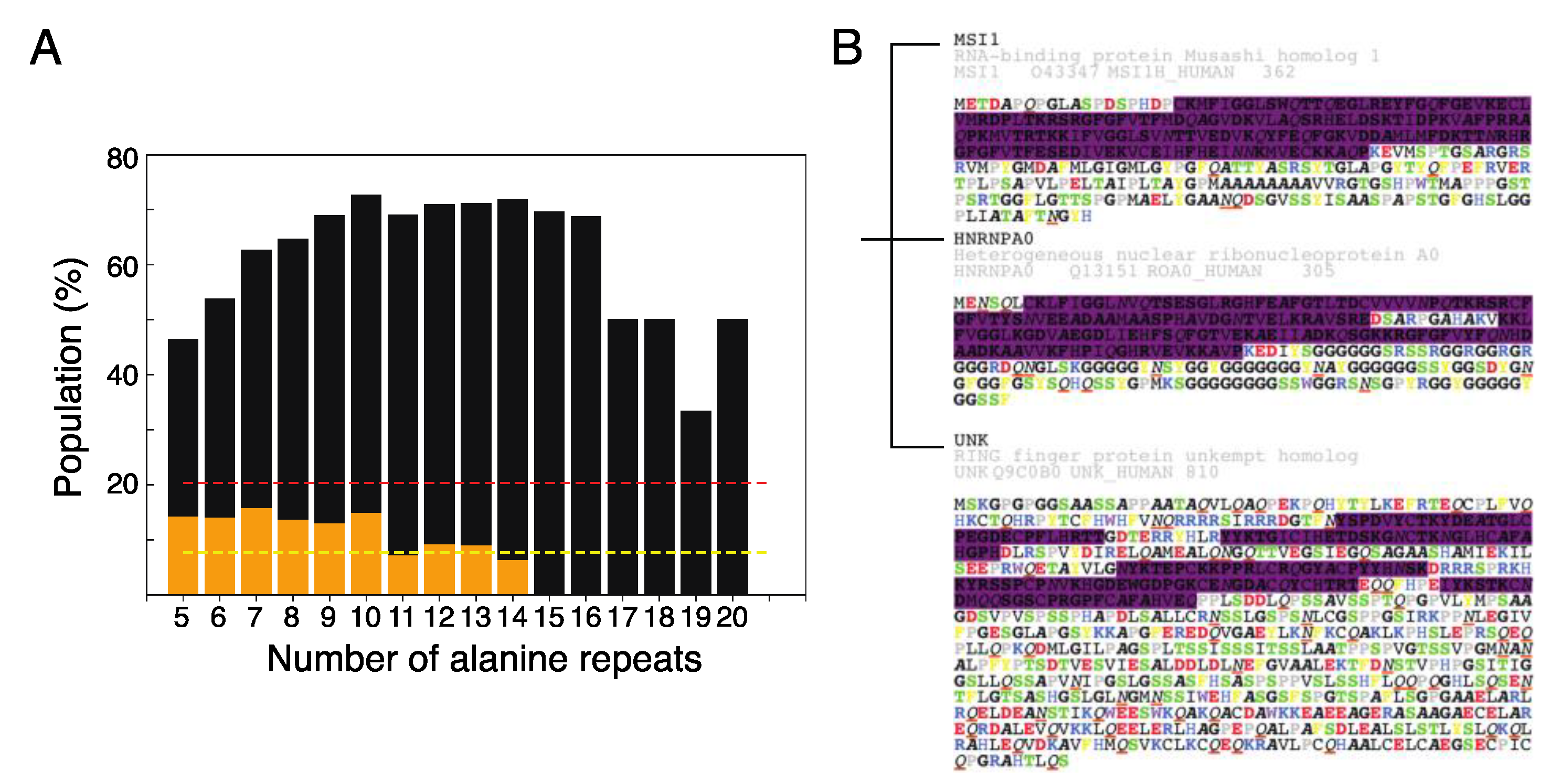
2.4. The Role of Polyalanine Tracts in Musashi-1 and in RBPs in General
3. Material and Methods
3.1. Musashi-1 Primary Sequence Analysis
3.2. Polyalanine Tracts Analysis
3.3. DNA Construct
3.4. Protein Expression and Purification
3.5. Chemical Shift Assignment
3.6. NMR Experiments and Analysis
3.7. Circular Dichroism Spectroscopy
3.8. Microscopy
3.9. Thioflavin T Fluorescence Assay
4. Conclusions
Accession Numbers
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RBP | RNA-binding proteins |
| IDP | Intrinsically disordered proteins |
| NMR | Nuclear magnetic resonance |
| CD | Circular dichroism |
| HSQC | Heteronuclear single-quantum coherence |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
References
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Zsolyomi, F.; Guharoy, M.; Tompa, P. Functional Advantages of Conserved Intrinsic Disorder in RNA-Binding Proteins. PLoS ONE 2015, 10, e0139731. [Google Scholar] [CrossRef]
- Zagrovic, B.; Bartonek, L.; Polyansky, A.A. RNA-protein interactions in an unstructured context. FEBS Lett. 2018, 592, 2901–2916. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Richard, S. Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem. Sci. 2015, 40, 662–672. [Google Scholar] [CrossRef]
- Jarvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubner, G.M.; Clery, A.; Allain, F.H. RRM-RNA recognition: NMR or crystallography... and new findings. Curr. Opin. Struct. Biol. 2013, 23, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Dorn, G.; Allain, F.H. Combining Mass Spectrometry (MS) and Nuclear Magnetic Resonance (NMR) Spectroscopy for Integrative Structural Biology of Protein-RNA Complexes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692.e20. [Google Scholar] [CrossRef] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef] [Green Version]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F.; et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 2018, 173, 720–734.e15. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Wang, X.J.; Vuong, C.K.; Lin, C.H.; Damianov, A.; Black, D.L. Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell 2017, 170, 312–323.e10. [Google Scholar] [CrossRef] [Green Version]
- Gueroussov, S.; Weatheritt, R.J.; O’Hanlon, D.; Lin, Z.Y.; Narula, A.; Gingras, A.C.; Blencowe, B.J. Regulatory Expansion in Mammals of Multivalent hnRNP Assemblies that Globally Control Alternative Splicing. Cell 2017, 170, 324–339.e23. [Google Scholar] [CrossRef] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.R.; Chen, T.C.; Hsiao, C.L.; Shi, L.; Chou, C.Y.; Huang, J.R. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta 2018, 1866, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Dignon, G.L.; Zerze, G.H.; Chabata, C.V.; Silva, R.; Conicella, A.E.; Amaya, J.; Burke, K.A.; Mittal, J.; Fawzi, N.L. Mechanistic View of hnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation. Mol. Cell 2018, 69, 465–479.e7. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.G.; Park, F.D.; Koechlein, C.S.; Kritzik, M.; Reya, T. Musashi signaling in stem cells and cancer. Annu. Rev. Cell Dev. Biol. 2015, 31, 249–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, U.; Montalbano, M.; McAllen, S.; Minuesa, G.; Kharas, M.; Kayed, R. Formation of Toxic Oligomeric Assemblies of RNA-binding Protein: Musashi in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbano, M.; McAllen, S.; Sengupta, U.; Puangmalai, N.; Bhatt, N.; Ellsworth, A.; Kayed, R. Tau oligomers mediate aggregation of RNA-binding proteins Musashi1 and Musashi2 inducing Lamin alteration. Aging Cell 2019, 18, e13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.F.; Wu, L.S.; Shen, C.K. TDP-43: An emerging new player in neurodegenerative diseases. Trends Mol. Med. 2008, 14, 479–485. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Tsai, K.L.; Huang, P.I.; Yang, Y.P.; Lee, Y.Y.; Chen, Y.W.; Lo, W.L.; Lan, Y.T.; et al. Musashi-1 promotes chemoresistant granule formation by PKR/eIF2alpha signalling cascade in refractory glioblastoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1850–1861. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Yang, T.W.; Huang, C.C.; Tang, C.Y.; Yen, J.Y.; Tsai, M.C.; Chen, H.Y.; Fadhilah, N.; Lin, C.C.; Jong, Y.J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef] [Green Version]
- Sigrist, C.J.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010, 38, D161–D166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwaoka, R.; Nagata, T.; Tsuda, K.; Imai, T.; Okano, H.; Kobayashi, N.; Katahira, M. Structural Insight into the Recognition of r(UAG) by Musashi-1 RBD2, and Construction of a Model of Musashi-1 RBD1-2 Bound to the Minimum Target RNA. Molecules 2017, 22, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, T.; Nagata, T.; Tsuda, K.; Kobayashi, N.; Imai, T.; Okano, H.; Yamazaki, T.; Katahira, M. Structure of Musashi1 in a complex with target RNA: The role of aromatic stacking interactions. Nucleic Acids Res. 2012, 40, 3218–3231. [Google Scholar] [CrossRef] [Green Version]
- Wootton, J.C. Non-globular domains in protein sequences: Automated segmentation using complexity measures. Comput. Chem. 1994, 18, 269–285. [Google Scholar] [CrossRef]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Obradović, Z.; Brown, C.J.; Dunker, A.K. Prediction of boundaries between intrinsically ordered and disordered protein regions. Pac. Symp. Biocomput. 2003, 8, 216–227. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Seno, F.; Tosatto, S.C.; Trovato, A. PASTA 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef]
- Camilloni, C.; De Simone, A.; Vranken, W.F.; Vendruscolo, M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 2012, 51, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Wang, L.; Ishikawa, R.; Li, Y.; Fiedler, M.; Liu, F.; Calder, G.; Rowan, B.; Weigel, D.; Li, P.; et al. Arabidopsis FLL2 promotes liquid-liquid phase separation of polyadenylation complexes. Nature 2019, 569, 265–269. [Google Scholar] [CrossRef]
- Brown, L.Y.; Brown, S.A. Alanine tracts: The expanding story of human illness and trinucleotide repeats. Trends Genet. 2004, 20, 51–58. [Google Scholar] [CrossRef]
- Li, L.; Ng, N.K.; Koon, A.C.; Chan, H.Y. Expanded polyalanine tracts function as nuclear export signals and promote protein mislocalization via eEF1A1 factor. J. Biol. Chem. 2017, 292, 5784–5800. [Google Scholar] [CrossRef] [Green Version]
- Polling, S.; Ormsby, A.R.; Wood, R.J.; Lee, K.; Shoubridge, C.; Hughes, J.N.; Thomas, P.Q.; Griffin, M.D.; Hill, A.F.; Bowden, Q.; et al. Polyalanine expansions drive a shift into alpha-helical clusters without amyloid-fibril formation. Nat. Struct. Mol. Biol. 2015, 22, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Itakura, A.K.; Futia, R.A.; Jarosz, D.F. It Pays To Be in Phase. Biochemistry 2018, 57, 2520–2529. [Google Scholar] [CrossRef]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, Y.; Kulminskaya, N.V.; Mulder, F.A. Easy and unambiguous sequential assignments of intrinsically disordered proteins by correlating the backbone 15N or 13C’ chemical shifts of multiple contiguous residues in highly resolved 3D spectra. J. Biomol. NMR 2015, 61, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef]
- Liu, M.; Mao, X.A.; Ye, C.; Huang, H.; Nicholson, J.K.; Lindon, J.C. Improved WATERGATE Pulse Sequences for Solvent Suppression in NMR Spectroscopy. J. Magn. Reson. 1998, 132, 125–129. [Google Scholar] [CrossRef]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Dubochet, J. On the Development of Electron Cryo-Microscopy (Nobel Lecture). Angew. Chem. Int. Ed. 2018, 57, 10842–10846. [Google Scholar] [CrossRef]
- Henderson, R. From Electron Crystallography to Single Particle CryoEM (Nobel Lecture). Angew. Chem. Int. Ed. 2018, 57, 10804–10825. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Single-Particle Reconstruction of Biological Molecules-Story in a Sample (Nobel Lecture). Angew. Chem. Int. Ed. 2018, 57, 10826–10841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Leiro, R.; Scheres, S.H. Unravelling biological macromolecules with cryo-electron microscopy. Nature 2016, 537, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, H.J.; Wright, P.E. Perspective: The essential role of NMR in the discovery and characterization of intrinsically disordered proteins. J. Biomol. NMR 2019, 73, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, H.; Imai, T.; Imataka, H.; Tsujimoto, M.; Matsumoto, K.; Okano, H. Neural RNA-binding protein Musashi1 inhibits translation initiation by competing with eIF4G for PABP. J. Cell Biol. 2008, 181, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Cragle, C.; MacNicol, A.M. Musashi protein-directed translational activation of target mRNAs is mediated by the poly(A) polymerase, germ line development defective-2. J. Biol. Chem. 2014, 289, 14239–14251. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-C.; Huang, J.-r. Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of RNA-Binding Proteins. Int. J. Mol. Sci. 2020, 21, 2289. https://doi.org/10.3390/ijms21072289
Chen T-C, Huang J-r. Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of RNA-Binding Proteins. International Journal of Molecular Sciences. 2020; 21(7):2289. https://doi.org/10.3390/ijms21072289
Chicago/Turabian StyleChen, Tsai-Chen, and Jie-rong Huang. 2020. "Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of RNA-Binding Proteins" International Journal of Molecular Sciences 21, no. 7: 2289. https://doi.org/10.3390/ijms21072289
APA StyleChen, T. -C., & Huang, J. -r. (2020). Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of RNA-Binding Proteins. International Journal of Molecular Sciences, 21(7), 2289. https://doi.org/10.3390/ijms21072289