Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment

 , ,
, , 
 and
and
Abstract
:1. Introduction
2. Results
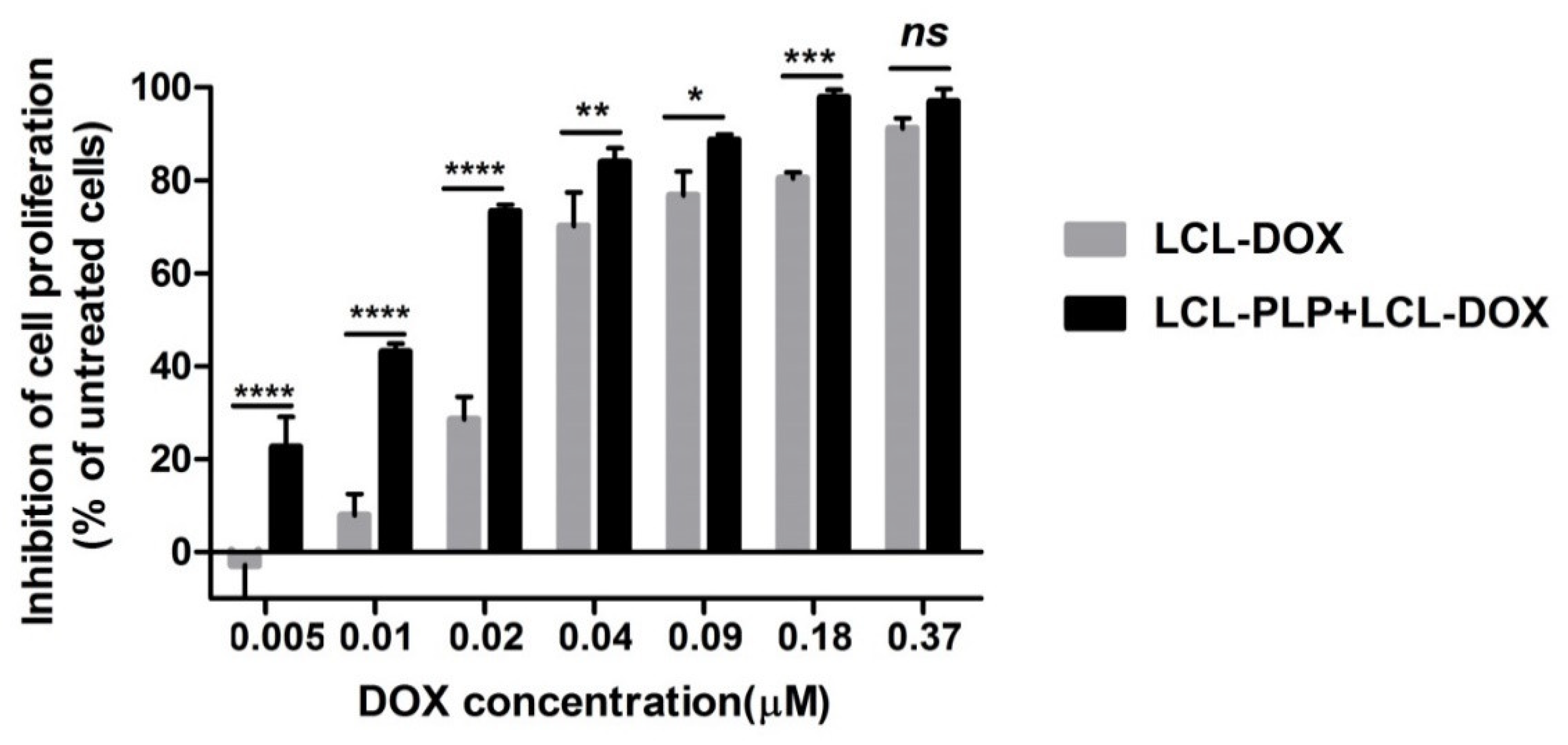
2.1. Effects of the Liposomal Combination Therapy on the Proliferation of B16.F10 Cell Co-Cultivated with Murine Macrophages
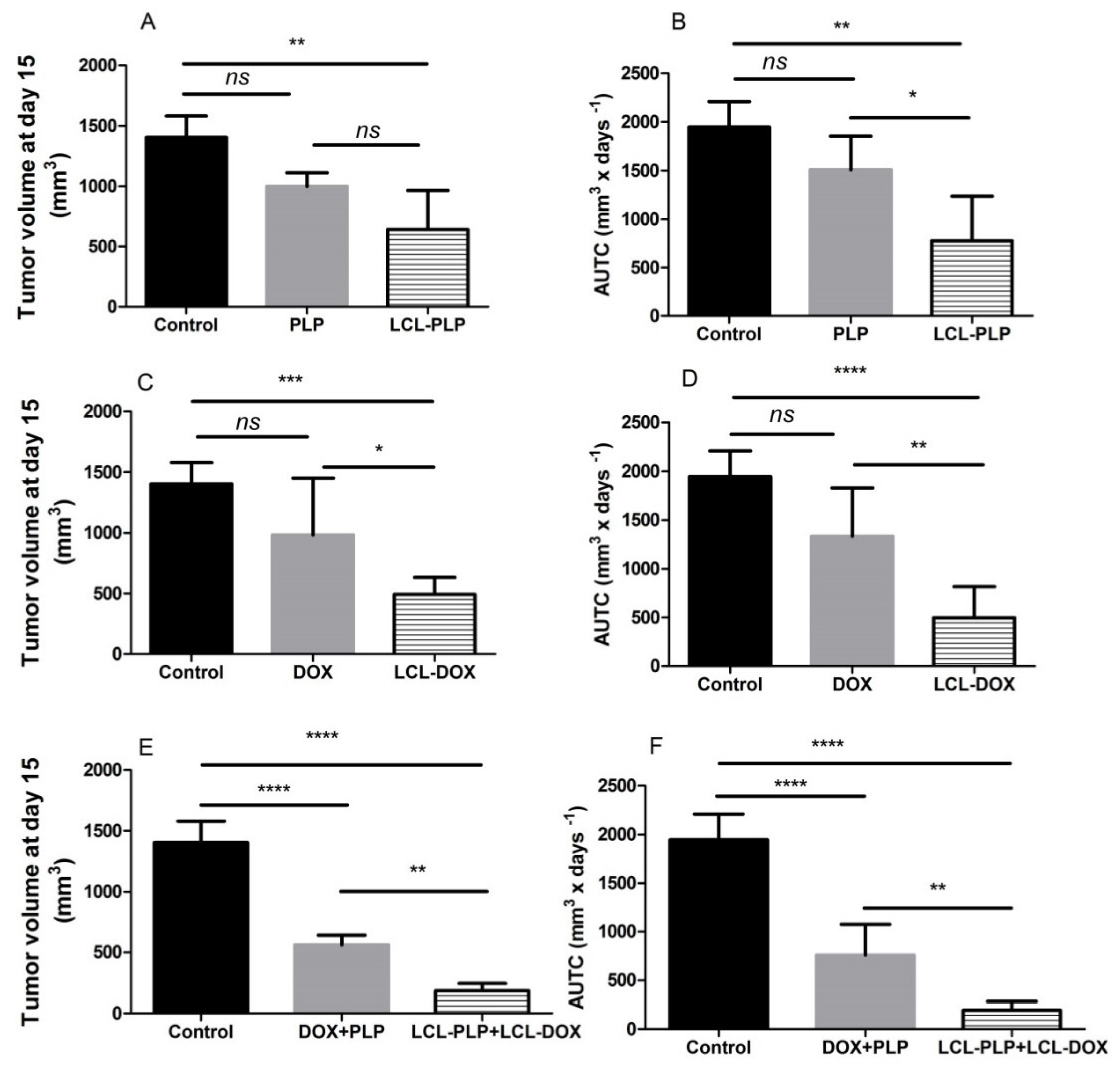
2.2. The Combined Liposomal Drug Therapy Induced a Stronger Inhibition of the Melanoma Tumor Growth than Monotherapies Based on either LCL-DOX or LCL-PLP
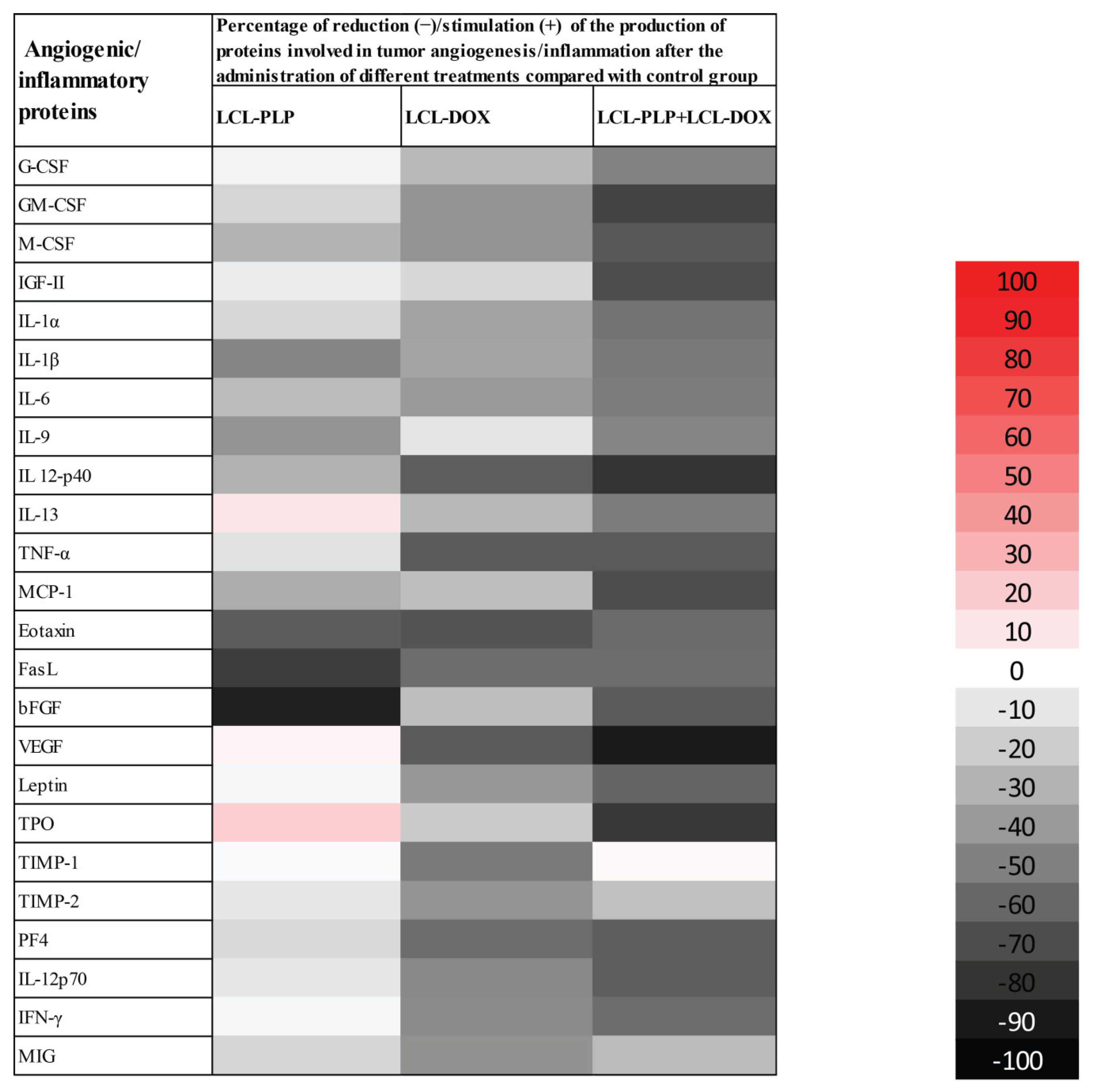
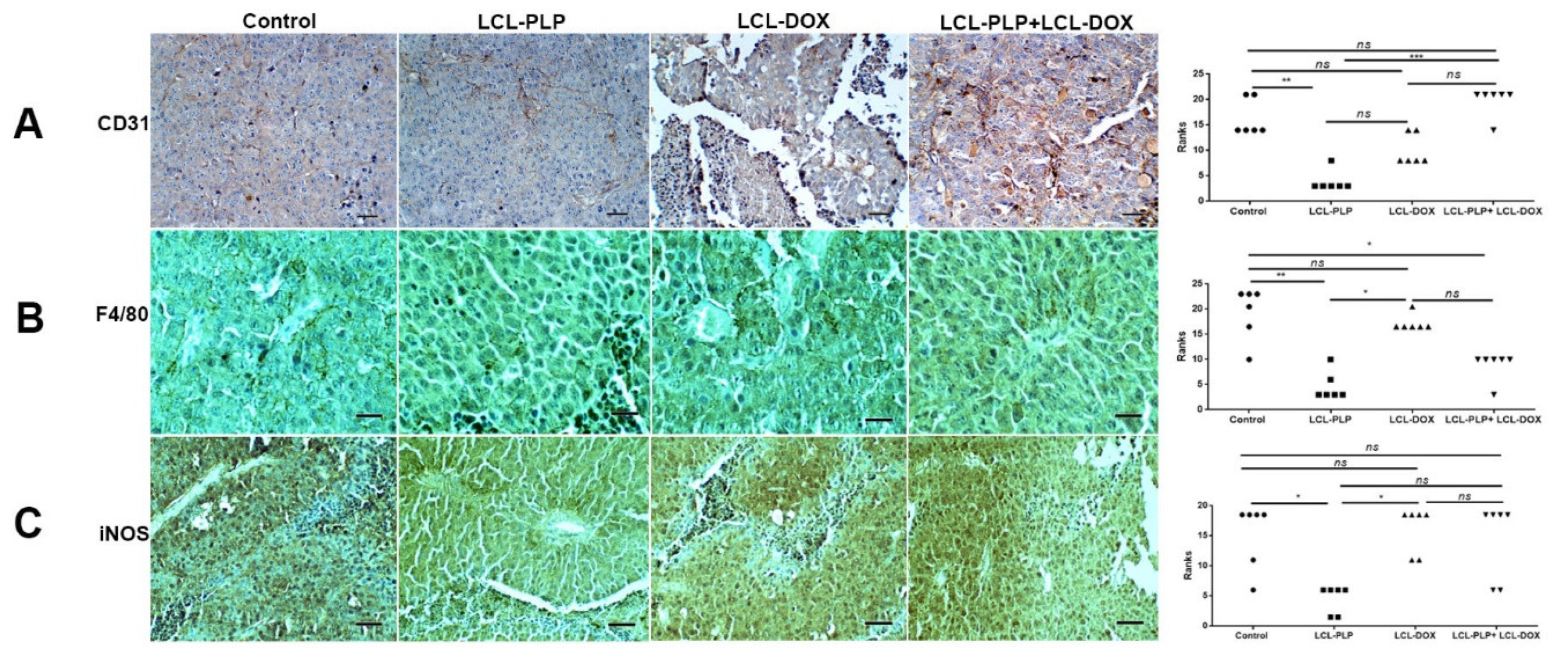
2.3. Liposomal Combination Therapy Induced Strong Anti-Angiogenic Actions on Melanoma in Vivo
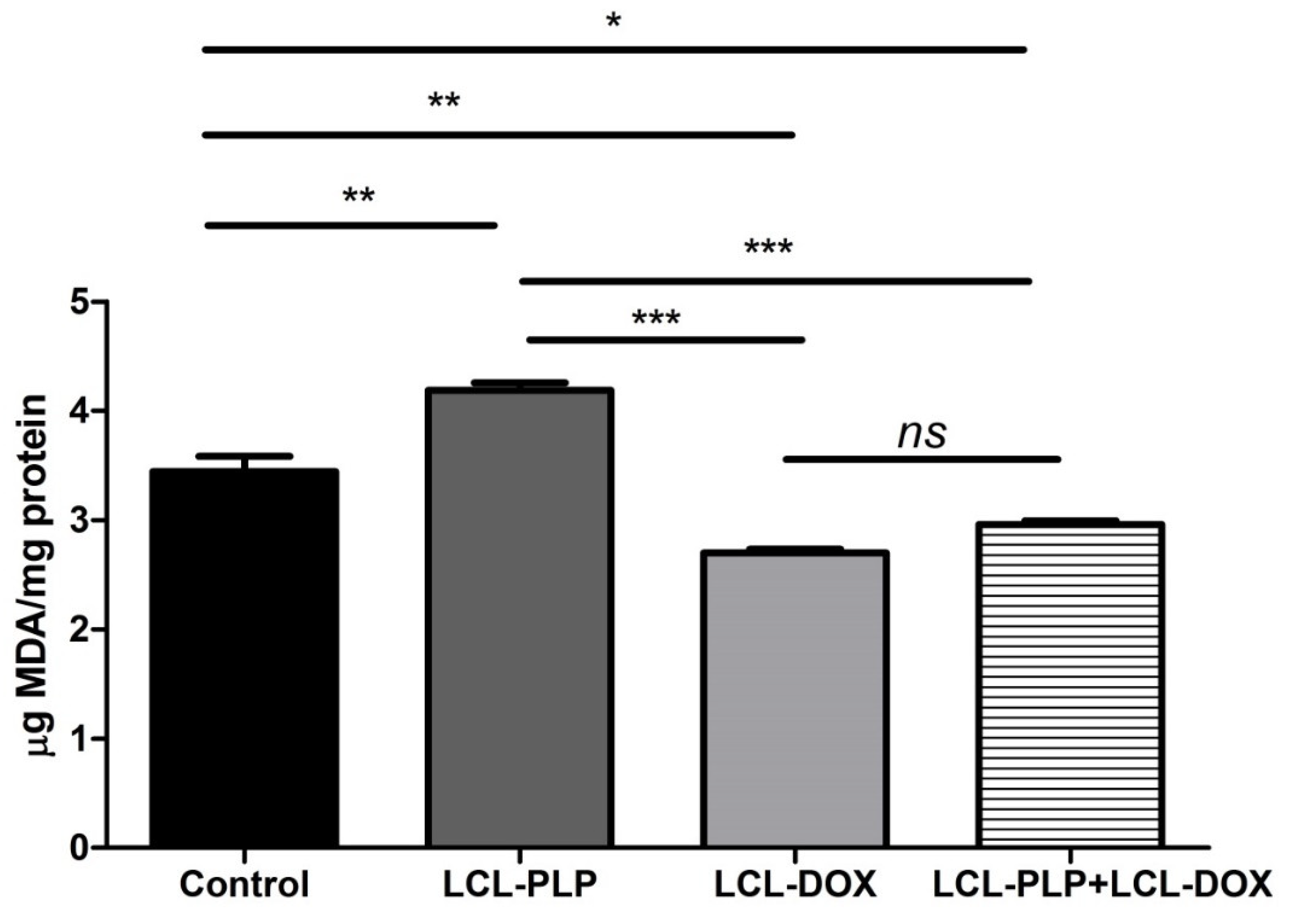
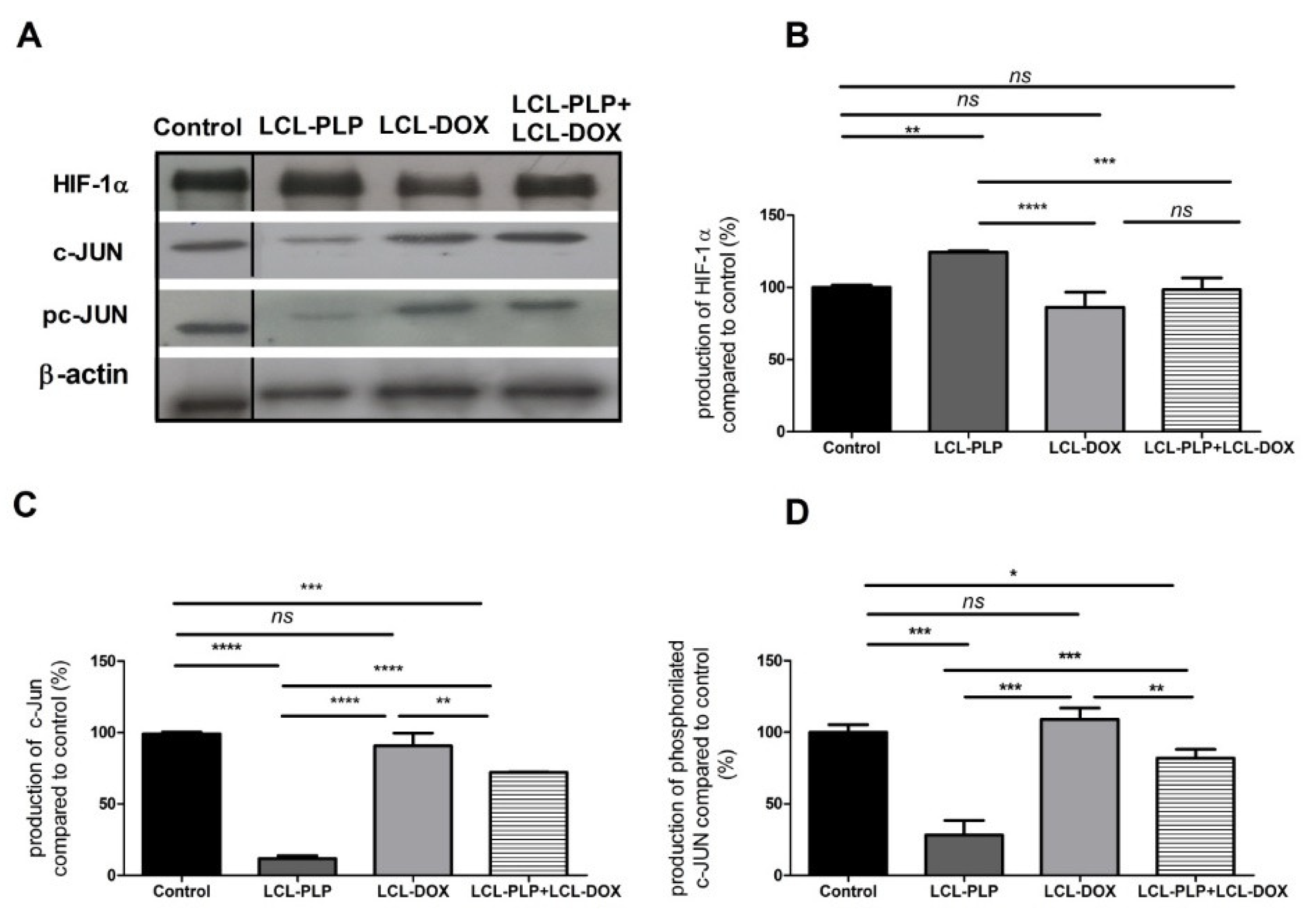
2.4. Combined Therapy Induced Slight Reduction of the Intratumor Oxidative Stress
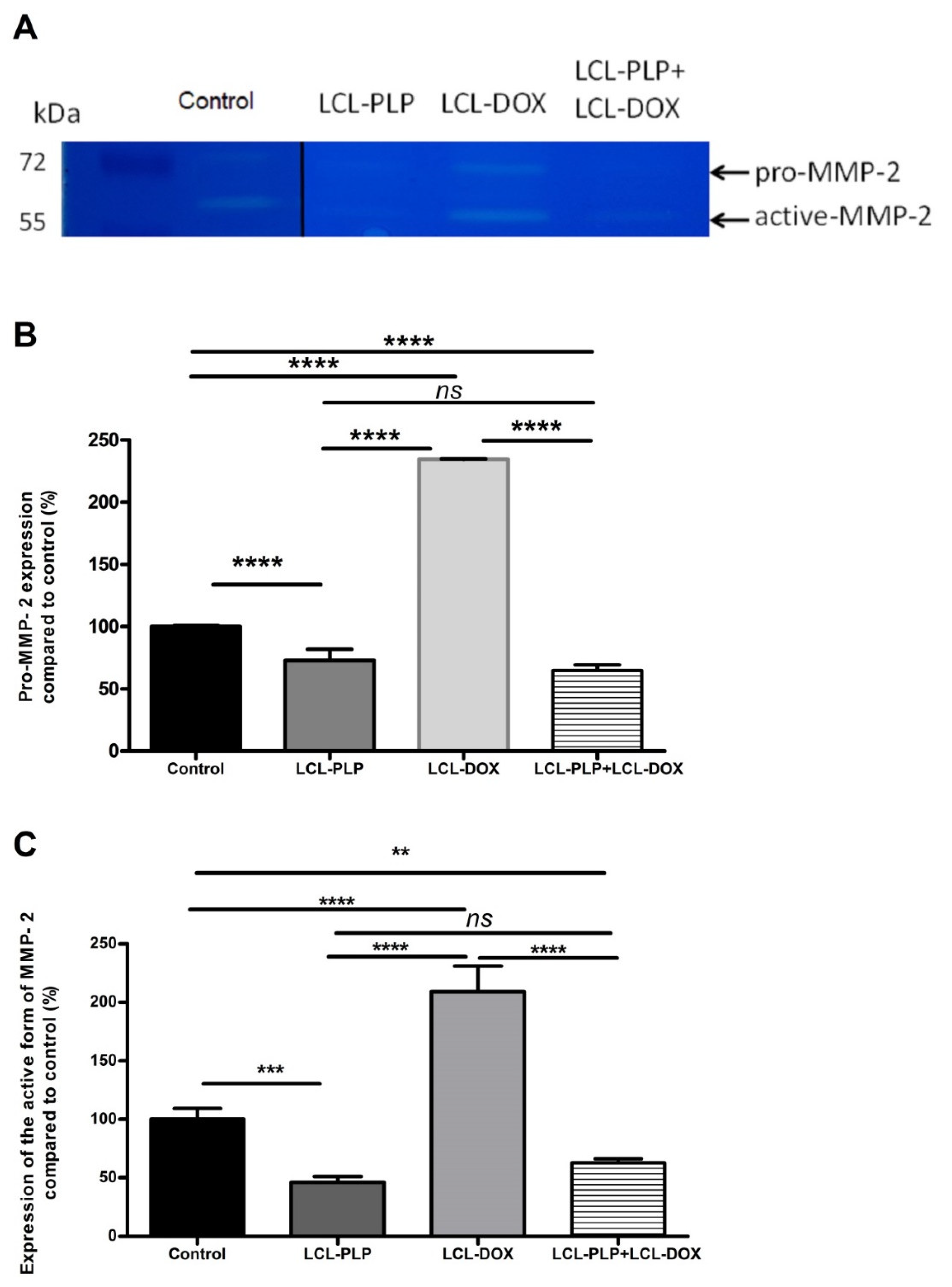
2.5. Combined Liposomal Drug Therapy Reduced the Aggressiveness of B16.F10 Melanoma in Vivo
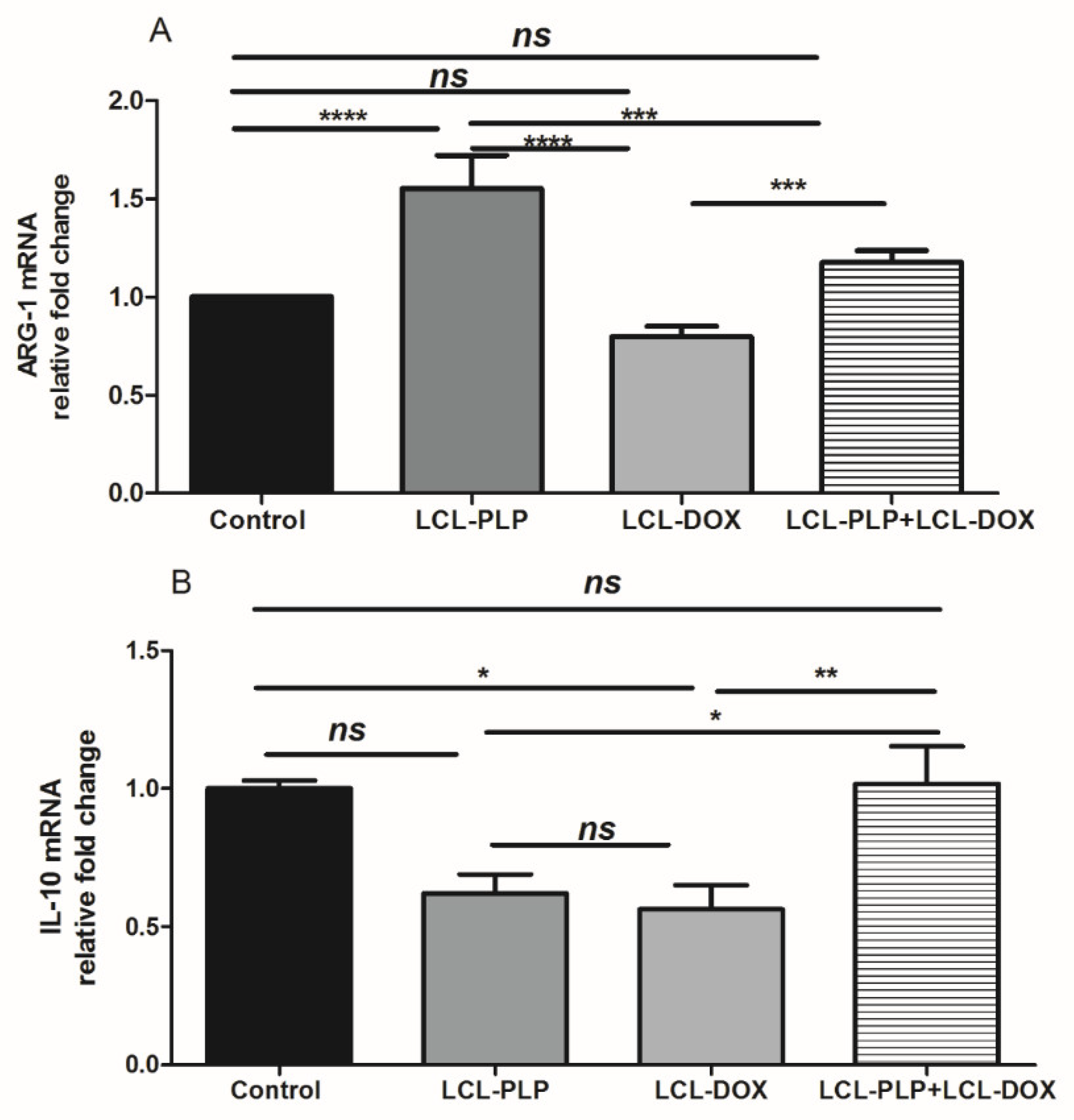
2.6. Combination Therapy Reduced Macrophage Density in Melanoma Microenvironment
3. Discussion
4. Materials and Methods
4.1. Preparation of Liposomal Formulations
4.2. Cell Types and Culture Conditions
4.3. In Vivo Melanoma Model
4.4. Evaluation of the Antiproliferative Activity of the Liposomal Combination Therapy on B16.F10 Cells
4.5. Effects of Combination Therapy (LCL-PLP + LCL-DOX) on Tumor Growth
4.6. Western Blot Quantification of the HIF-1α and c-Jun Levels in Tumor Tissue
4.7. Protein Array Analysis of the Inflammatory/Angiogenic Protein Levels in Tumors
4.8. HPLC Determination of Malondialdehyde Levels in Tumor Cell Lysates
4.9. RT-qPCR Determination of Arginase-1 and IL-10 Expression
4.10. Determination of MMP-2 Activity by Gelatin Zymography
4.11. Immunohistochemistry Analysis of Tumor Tissue
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| Arg-1 | arginase-1 |
| bFGF BMDMs | Basic fibroblast growth factor Bone marrow-derived macrophages; |
| BrdU | bromodeoxyuridine |
| DMEM | Dulbecco’s Modified Eagle’s medium |
| DOX | doxorubicin |
| FasL | Fas ligand |
| G-CSF | Granulocyte-colony stimulating factor |
| GM-CSF | Granulocyte-macrophage-colony stimulating factor |
| HPLC | high-performance liquid chromatography |
| IC50 | Half maximal inhibitory concentration |
| IFN-γ | Interferon γ |
| IGF-II | Insulin growth factor II |
| IL-1α | Interleukin 1α |
| IL-1β | Interleukin 1β |
| IL-4 | Interleukin 4 |
| IL-6 | Interleukin 6 |
| IL-9 | Interleukin 9 |
| IL-10 | Interleukin 10 |
| IL-12 p40 | Interleukin 12 p40 |
| IL-12 p70 | Interleukin 12 p70 |
| IL-13 | Interleukin 13 |
| MDA | Malondialdehyde |
| MIG | Monokine induced by IFN-γ |
| M-CSF | Monocyte-colony stimulating factor |
| MCP-1 | Monocyte chemoattractant protein-1 |
| PF-4 | Platelet factor 4 |
| PLP | Prednisolone disodium phosphate |
| SD | Standard deviation |
| TAMs | tumor - associated macrophages |
| TBS | Tris-buffered saline |
| TIMP-1 | Tissue inhibitor of metalloproteinase 1 |
| TIMP-2 | Tissue inhibitor of metalloproteinase 2 |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor α |
| TPO | Thrombopoietin |
| VEGF | Vascular endothelial growth factor |
References
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Carè, A.; Matarrese, P. Cell death-based treatments of melanoma:conventional treatments and new therapeutic strategies. Cell Death Dis. 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef]
- Somasundaram, R.; Herlyn, M.; Wagner, S. The role of tumor microenvironment in melanoma therapy resistance. Melanoma Manag. 2016, 3, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, L.; Wang, N.; Zhang, Q.; Zhang, L. Pro-tumor activities of macrophages in the progression of melanoma. Hum. Vaccines Immunother. 2017, 13, 1556–1562. [Google Scholar] [CrossRef]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Deng, G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018, 7, 81–87. [Google Scholar]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Banciu, M.; Metselaar, J.M.; Schiffelers, R.M.; Storm, G. Antitumor Activity of Liposomal Prednisolone Phosphate Depends on the Presence of Functional Tumor-Associated Macrophages in Tumor Tissue. Neoplasia 2008, 10, 108–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smylie, M.G.; Wong, R.; Mihalcioiu, C.; Lee, C.; Pouliot, J.-F. A phase II, open label, monotherapy study of liposomal doxorubicin in patients with metastatic malignant melanoma. Investig. New Drugs 2006, 25, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, M.; Schadendorf, D.; Fink, W.; Zimpfer-Rechner, C.; Thoelke, A.; Figl, R.; Kaatz, M. Clinical Phase II Study of Pegylated Liposomal Doxorubicin as Second-Line Treatment in Disseminated Melanoma. Oncol. Res. Treat. 2004, 27, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Licarete, E.; Rauca, V.F.; Luput, L.; Patras, L.; Sesarman, A.; Banciu, M. The prednisolone phosphate-induced suppression of the angiogenic function of tumor-associated macrophages enhances the antitumor effects of doxorubicin on B16.F10 murine melanoma cells in vitro. Oncol. Rep. 2019, 42, 2694–2705. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Venza, M.; Visalli, M.; Beninati, C.; De Gaetano, G.V.; Teti, D.; Venza, I. Cellular Mechanisms of Oxidative Stress and Action in Melanoma. Oxidative Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cannavò, S.P.; Tonacci, A.; Bertino, L.; Casciaro, M.; Borgia, F.; Gangemi, S.; Casciaro, M.; Borgia, F. The role of oxidative stress in the biology of melanoma: A systematic review. Pathol.—Res. Pract. 2019, 215, 21–28. [Google Scholar] [CrossRef]
- Qutub, A.A.; Popel, A.S. Reactive Oxygen Species Regulate Hypoxia-Inducible Factor 1α Differentially in Cancer and Ischemia. Mol. Cell. Boil. 2008, 28, 5106–5119. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, U.B.; Westphal, J.R.; Waas, E.T.; Zendman, A.J.W.; Cornelissen, I.M.H.A.; Ruiter, D.J.; Van Muijen, G.N.P. Matrix metalloproteinases in human melanoma cell lines and xenografts: Increased expression of activated matrix metalloproteinase-2 (MMP-2) correlates with melanoma progression. Br. J. Cancer 1999, 81, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Li, H.; Zhang, Y.; Yang, H.; Guo, M.; Li, L.; Liu, T. Matrix metalloproteinase 2 promotes cell growth and invasion in colorectal cancer. Acta Biochim. et Biophys. Sin. 2011, 43, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Bergman, M.R.; Cheng, S.; Honbo, N.; Piacentini, L.; Karliner, J.S.; Lovett, D.H. A functional activating protein 1 (AP-1) site regulates matrix metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1 and JunB-FosB heterodimers. Biochem. J. 2003, 369, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.N.; Joshi, S.S.; Niles, R.M. Expression and function of hypoxia inducible factor-1 alpha in human melanoma under non-hypoxic conditions. Mol. Cancer 2009, 8, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, A.M.; Hermanns, M.; Skrzynski, C.; Nesslinger, M.; Muller, K.-M.; Kirkpatrick, C. Expression of the Endothelial Markers PECAM-1, vWf, and CD34 in Vivo and in Vitro. Exp. Mol. Pathol. 2002, 72, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Jürgensen, H.J.; Siersbæk, M.S.; Kuczek, D.E.; Cloud, L.G.; Liu, S.; Behrendt, N.; Grøntved, L.; Weigert, R.; Bugge, T.H. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017, 21, 3662–3671. [Google Scholar] [CrossRef] [Green Version]
- Weisser, S.B.; McLarren, K.W.; Kuroda, E.; Sly, L. Generation and Characterization of Murine Alternatively Activated Macrophages. Adv. Struct. Saf. Stud. 2012, 946, 225–239. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F. Identifying and Overcoming Immune Resistance Mechanisms in the Melanoma Tumor Microenvironment. Clin. Cancer Res. 2006, 12, 2326–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediat. Inflamm. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.; Elie, B.T.; Kester, J.C.; Garfall, A.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, O.; Kim, H.; Quail, D.F.; Foley, E.A.; Joyce, J.A. Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell Rep. 2017, 19, 101–113. [Google Scholar] [CrossRef] [PubMed]
- A Vorobiof, D.; Rapoport, B.L.; Mahomed, R.; Karime, M. Phase II study of pegylated liposomal doxorubicin in patients with metastatic malignant melanoma failing standard chemotherapy treatment. Melanoma Res. 2003, 13, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Banciu, M.; Schiffelers, R.M.; Storm, G. Investigation into the Role of Tumor-Associated Macrophages in the Antitumor Activity of Doxil. Pharm. Res. 2008, 25, 1948–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyndam, M.C.; Van Berkel, M.P.; Dorsman, J.C.; Rockx, D.A.; Pinedo, H.M.; Boven, E. Cisplatin and doxorubicin repress Vascular Endothelial Growth Factor expression and differentially down-regulate Hypoxia-inducible Factor I activity in human ovarian cancer cells. Biochem. Pharmacol. 2007, 74, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Ogata, S.; Yorioka, N.; Kohno, N. Glucose and prednisolone alter basic fibroblast growth factor expression in peritoneal mesothelial cells and fibroblasts. J. Am. Soc. Nephrol. 2001, 12, 2787–2796. [Google Scholar]
- Mrad, M.; Imbert, C.; Garcia, V.; Rambow, F.; Therville, N.; Carpentier, S.; Ségui, B.; Levade, T.; Azar, R.; Marine, J.-C.; et al. Downregulation of sphingosine kinase-1 induces protective tumor immunity by promoting M1 macrophage response in melanoma. Oncotarget 2016, 7, 71873–71886. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumor-Associated Macrophages: Therapeutic Targets for Skin Cancer. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.T.; Barnhill, R.; Galanopoulos, T.; Antoniades, H.N. Expression of monocyte chemotactic protein-1 in human melanoma in vivo. Am. J. Pathol. 1992, 140, 9–14. [Google Scholar]
- Lee, H.-W.; Choi, H.-J.; Ha, S.-J.; Lee, K.-T.; Kwon, Y.-G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. et Biophys. Acta (BBA)—Bioenerg. 2013, 1835, 170–179. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, X.; Zhu, L.; Li, S.; Xiao, Q.; He, W.; Yin, L. Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics 2018, 8, 2830–2845. [Google Scholar] [CrossRef]
- Tiago, M.; De Oliveira, E.; Brohem, C.A.; Pennacchi, P.; Paes, R.D.; Haga, R.B.; Campa, A.; Barros, S.B.D.M.; Smalley, K.S.; Maria-Engler, S.S. Fibroblasts Protect Melanoma Cells from the Cytotoxic Effects of Doxorubicin. Tissue Eng. Part A 2014, 20, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbel, M.; Lagente, V.; Théret, N.; Germain, N.; Clement, B.; Boichot, E. Comparative Effects of Betamethasone, Cyclosporin and Nedocromil Sodium in Acute Pulmonary Inflammation and Metalloproteinase Activities in Bronchoalveolar Lavage Fluid from Mice Exposed to Lipopolysaccharide. Pulm. Pharmacol. Ther. 1999, 12, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Meier-Schiesser, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuphal, S.; Winklmeier, A.; Warnecke, C.; Bosserhoff, A. Constitutive HIF-1 activity in malignant melanoma. Eur. J. Cancer 2010, 46, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Vettori, A.; Greenald, D.; Wilson, G.K.; Peron, M.; Facchinello, N.; Markham, E.; Sinnakaruppan, M.; Matthews, L.C.; A McKeating, J.; Argenton, F.; et al. Glucocorticoids promote Von Hippel Lindau degradation and Hif-1α stabilization. Proc. Natl. Acad. Sci. USA 2017, 114, 9948–9953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, N.; Maruhashi, T.; Sugiura, D.; Shimizu, K.; Okazaki, I.-M.; Okazaki, T. Glucocorticoids potentiate the inhibitory capacity of programmed cell death 1 by up-regulating its expression on T cells. J. Boil. Chem. 2019, 294, 19896–19906. [Google Scholar] [CrossRef]
- Kim, D.J.; Jang, J.H.; Ham, S.-Y.; Choi, S.H.; Park, S.S.; Jeong, S.Y.; Kim, B.C.; Jeon, D.Y.; Lee, B.J.; Ko, B.K.; et al. Doxorubicin inhibits PD-L1 expression by enhancing TTP-mediated decay of PD-L1 mRNA in cancer cells. Biochem. Biophys. Res. Commun. 2019, 522, 402–407. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; Van De Vijver, K.; De Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Bolotin, E.M.; Cohen, R.; Bar, L.K.; Emanuel, N.; Ninio, S.; Barenholz, Y.; Lasic, D.D. Ammonium Sulfate Gradients for Efficient and Stable Remote Loading of Amphipathic Weak Bases into Liposomes and Ligandoliposomes. J. Liposome Res. 1994, 4, 455–479. [Google Scholar] [CrossRef]
- Licarete, E.; Sesarman, A.; Rauca, V.F.; Luput, L.; Patras, L.; Banciu, M. HIF-1α acts as a molecular target for simvastatin cytotoxicity in B16.F10 melanoma cells cultured under chemically induced hypoxia. Oncol. Lett. 2017, 13, 3942–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauca, V.-F.; Licarete, E.; Luput, L.; Sesarman, A.; Patras, L.; Bulzu, P.-A.; Rakosy-Tican, E.; Banciu, M. Combination therapy of simvastatin and 5, 6-dimethylxanthenone-4-acetic acid synergistically suppresses the aggressiveness of B16.F10 melanoma cells. PLoS ONE 2018, 13, e0202827. [Google Scholar] [CrossRef]
- Luput, L.; Licarete, E.; Drotar, D.M.; Nagy, A.-L.; Sesarman, A.; Patras, L.; Rauca, V.F.; Porfire, A.; Muntean, D.M.; Achim, M.; et al. In Vivo Double Targeting of C26 Colon Carcinoma Cells and Microenvironmental Protumor Processes Using Liposomal Simvastatin. J. Cancer 2018, 9, 440–449. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.; Fridman, R. Assessment of Gelatinases (MMP-2 and MMP-9 by Gelatin Zymography. Methods Mol. Med. 2001, 57, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angiogenic/Inflammatory Proteins | Percentage of Reduction (−)/Increase (+) in Intratumor Production of Proteins Involved in Tumor Angiogenesis/Inflammation Following Different Treatments Compared Their Levels in Control Tumors | ||
|---|---|---|---|
| LCL−PLP | LCL−DOX | LCL−PLP + LCL−DOX | |
| Granulocyte-colony stimulating factor (G-CSF) | −5.96 ± 4.03(ns) | −27.52 ± 3.93(**) | −49.73 ± 2.68(****) |
| Granulocyte−macrophage-colony stimulating factor (GM-CSF) | −16.80 ± 12.03 (ns) | −42.57 ± 4.35 (****) | −73.93 ± 0.94(****) |
| Monocyte−colony stimulating factor (M-CSF) | −30.00 ± 23.88(**) | −42.54 ± 2.81 (****) | −65.54 ± 4.04(****) |
| Insulin growth factor II (IGF-II) | −8.15 ± 24.81(ns) | −15.67 ± 2.14 (ns) | −70.12 ± 1.96 (***) |
| Interleukin 1α (IL-1α) | −15.52 ± 14.06(ns) | −36.20 ± 0.74(****) | −55.02 ± 1.93(****) |
| Interleukin 1β (IL-1β) | −48.17 ± 14.25(****) | −35.07 ± 3.80(***) | −53.58 ± 1.16(****) |
| Interleukin 6 (IL-6) | −26.49 ± 4.67(**) | −39.77 ± 7.08(****) | −52.05 ± 6.81(****) |
| Interleukin 9 (IL-9) | −42.70 ± 3.83(****) | −10.81 ± 1.53(ns) | −48.24 ± 5.69(****) |
| Interleukin 12 p40 (IL 12-p40) | −31.33 ± 0.35(***) | −63.90 ± 2.76(****) | −80.16 ± 0.07(****) |
| Interleukin 13 (IL-13) | +9.84 ± 5.91(ns) | −28.10 ± 1.29(***) | −51.88 ± 4.43(****) |
| Tumor necrosis factor α (TNF-α) | −12.42 ± 33.15(ns) | −64.83 ± 6.68(****) | −65.47 ± 0.00(****) |
| Monocyte chemoattractant protein-1 (MCP-1) | −32.62 ± 4.46(***) | −25.40 ± 5.37(*) | −69.89 ± 1.95(****) |
| Eotaxin | −64.09 ± 48.39(****) | −67.59 ± 1.33(****) | −58.79 ± 1.17(****) |
| Fas ligand (FasL) | −76.62 ± 17.48(****) | −57.25 ± 0.00(****) | −57.98 ± 5.15(****) |
| Basic fibroblast growth factor (bFGF) | −87.15 ± 4.06(****) | −25.44 ± 9.35(*) | −64.86 ± 0.60(****) |
| Vascular endothelial growth factor (VEGF) | +5.32 ± 63.80(ns) | −64.91 ± 19.15(****) | −89.14 ± 12.10(****) |
| Leptin | −4.78 ± 6.36(ns) | −40.51 ± 4.45(****) | −60.75 ± 15.09(****) |
| Thrombopoietin (TPO) | +19.46 ± 3.42(ns) | −20.31 ± 13.03(ns) | −78.09 ± 2.80(****) |
| Tissue inhibitor of matrix metalloproteinase 1 (TIMP-1) | −2.52 ± 10.78(ns) | −52.59 ± 0.87(****) | +3.76 ± (ns) |
| Tissue inhibitor of matrix metalloproteinase 2 (TIMP-2) | −10.02 ± 10.57(ns) | −42.47 ± 19.90(****) | −24.96 ± 21.18(*) |
| Platelet factor 4 (PF4) | −15.18 ± 2.11(ns) | −57.68 ± 3.72(****) | −58.17 ± 8.63(****) |
| Interleukin 12 p70 (IL-12p70) | −10.02 ± 10.57(ns) | −46.74 ± 1.08(****) | −63.45 ± 2.07(****) |
| Interferon γ (IFN-γ) | −4.69 ± 0.42(ns) | −45.92 ± 4.58(****) | −57.69 ± 7.82(****) |
| Monokine induced by IFN-γ (MIG) | −15.82 ± 1.25(ns) | −42.85 ± 33.18(****) | −26.30 ± 37.68(**) |
| Name of Genes | Forward Primer (5′-3′) | Reverse Primer(5′-3′) |
|---|---|---|
| Mouse β-actin | TCTTTGCAGCTCCTTCGTTGCCGGTCC | GTCCTTCTGACCCATTCCCACCATCACAC |
| Mouse Arg-1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Mouse IL-10 | GGTTGCCAAGCCTTATCGGA | ACCTGCTCCACTGCCTTGCT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Licarete, E.; Rauca, V.F.; Luput, L.; Drotar, D.; Stejerean, I.; Patras, L.; Dume, B.; Toma, V.A.; Porfire, A.; Gherman, C.; et al. Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 2968. https://doi.org/10.3390/ijms21082968
Licarete E, Rauca VF, Luput L, Drotar D, Stejerean I, Patras L, Dume B, Toma VA, Porfire A, Gherman C, et al. Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment. International Journal of Molecular Sciences. 2020; 21(8):2968. https://doi.org/10.3390/ijms21082968
Chicago/Turabian StyleLicarete, Emilia, Valentin Florian Rauca, Lavinia Luput, Denise Drotar, Ioana Stejerean, Laura Patras, Bogdan Dume, Vlad Alexandru Toma, Alina Porfire, Claudia Gherman, and et al. 2020. "Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment" International Journal of Molecular Sciences 21, no. 8: 2968. https://doi.org/10.3390/ijms21082968
APA StyleLicarete, E., Rauca, V. F., Luput, L., Drotar, D., Stejerean, I., Patras, L., Dume, B., Toma, V. A., Porfire, A., Gherman, C., Sesarman, A., & Banciu, M. (2020). Overcoming Intrinsic Doxorubicin Resistance in Melanoma by Anti-Angiogenic and Anti-Metastatic Effects of Liposomal Prednisolone Phosphate on Tumor Microenvironment. International Journal of Molecular Sciences, 21(8), 2968. https://doi.org/10.3390/ijms21082968