
Treg-Resistant Cytotoxic CD4+ T Cells Dictate T Helper Cells in Their Vicinity: TH17 Skewing and Modulation of Proliferation

 and
and
Abstract
:1. Introduction
2. Results
2.1. CD4 CTL Resist Treg-Mediated Suppression Independent of Their Expression of IL-10R and GITR
2.2. Conditioned Medium of CD4 CTL Enhances the Suppressive Phenotype of Tregs, but Decreases Functional Suppression of Naïve TH Cells
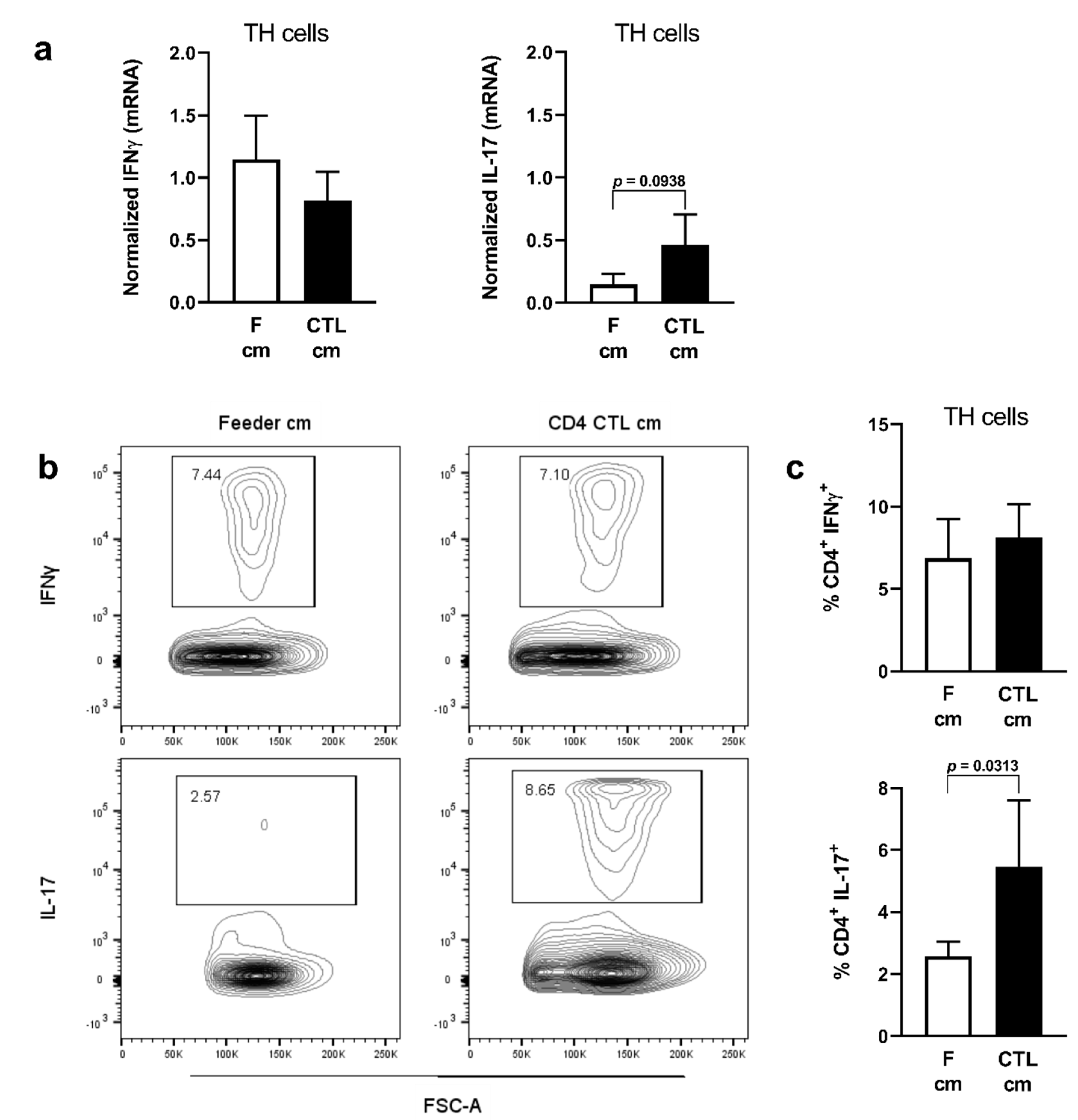
2.3. Conditioned Medium of CD4 CTL Promotes IL-17 Production
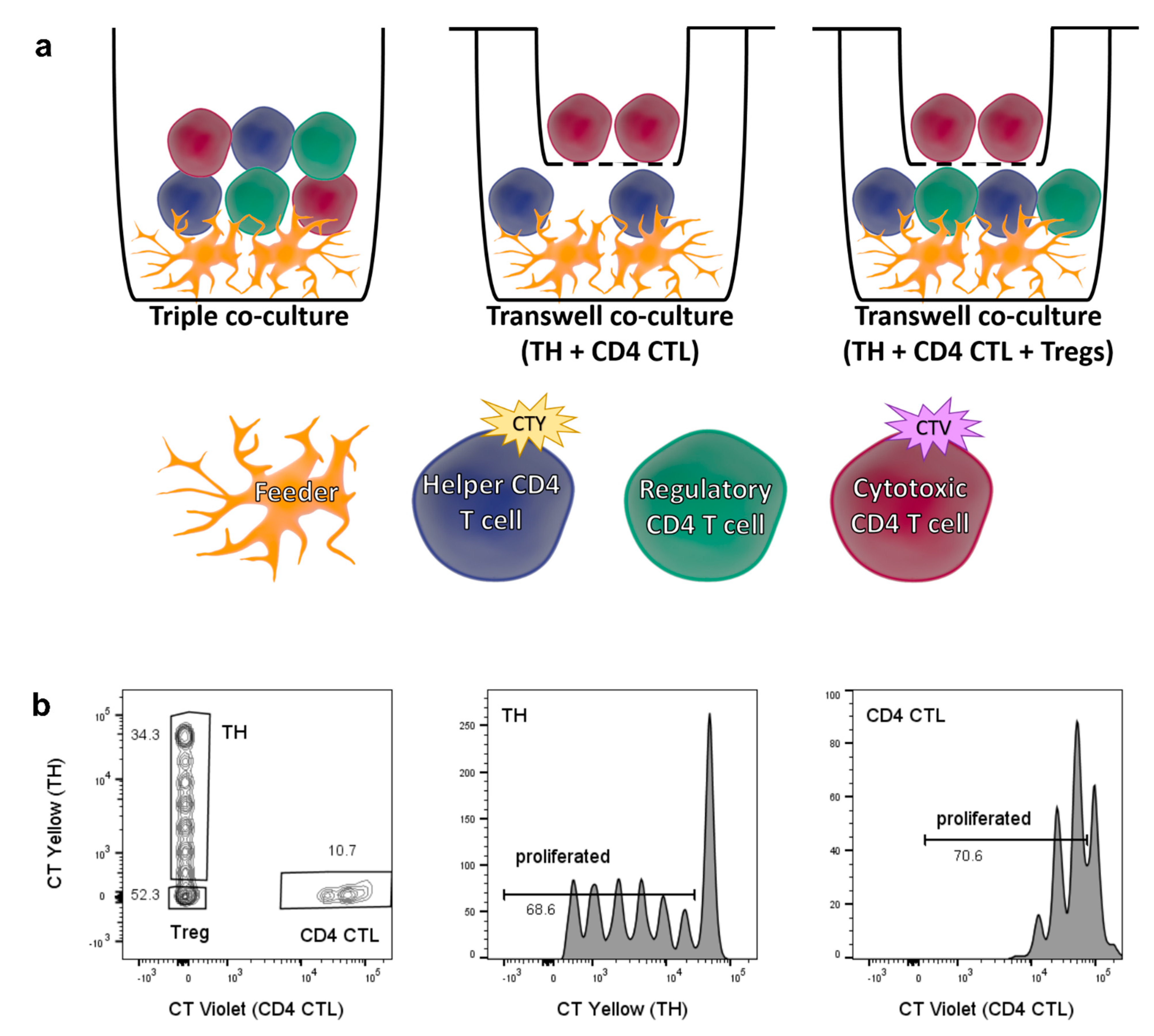
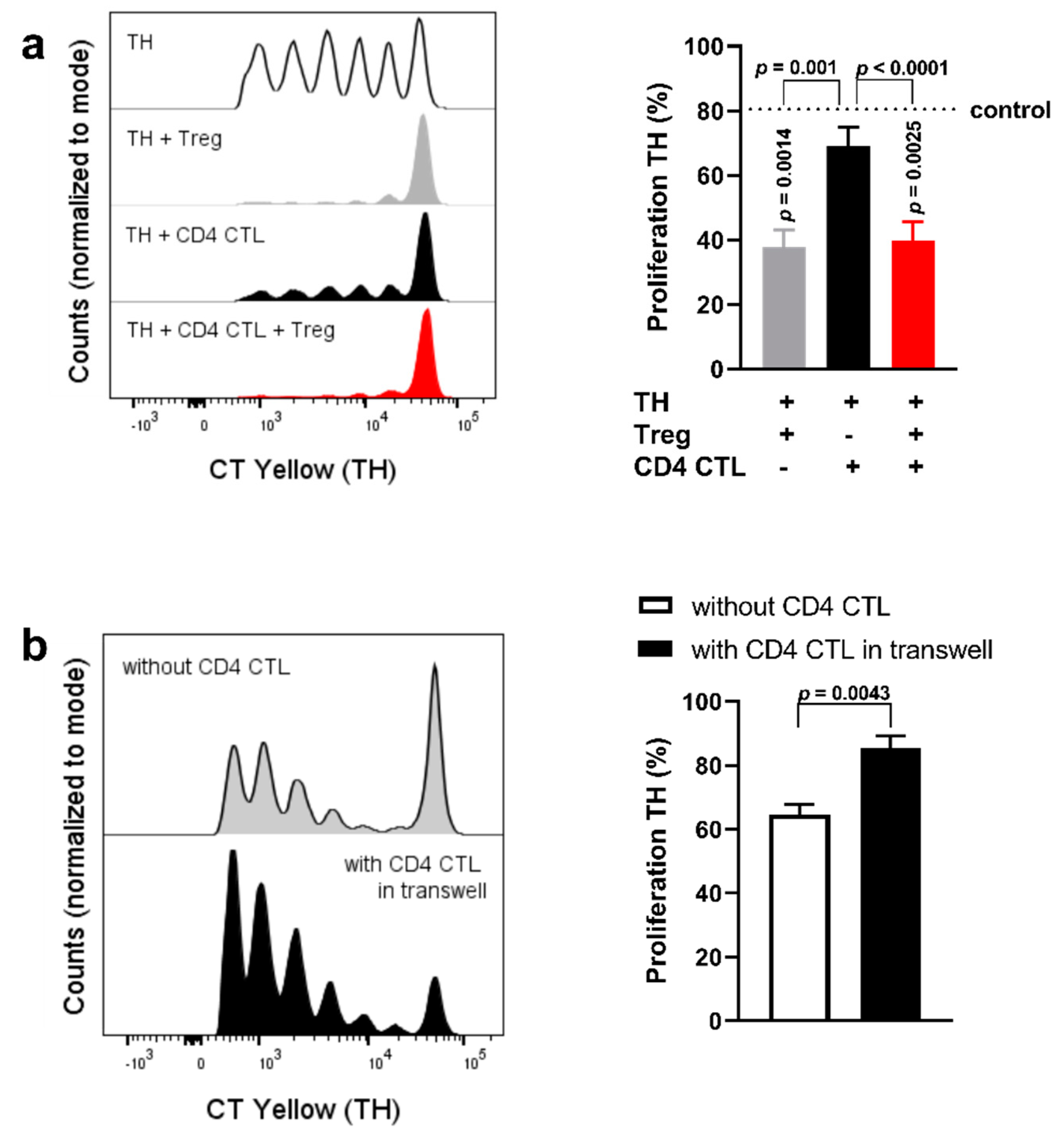
2.4. Cell−Cell Interaction between CD28+ TH Cells and CD4 CTL Reverts the Proliferation-Inducing Effects of the CD4 CTL Secretome
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chamberlain, W.D.; Falta, M.T.; Kotzin, B.L. Functional subsets within clonally expanded CD8+ memory T cells in elderly humans. Clin. Immunol. 2000, 94, 160–172. [Google Scholar] [CrossRef]
- Effros, R.B. Loss of CD28 expression on T lymphocytes: A marker of replicative senescence. Dev. Comp. Immunol. 1997, 21, 471–478. [Google Scholar] [CrossRef]
- Fiorentini, S.; Malacarne, F.; Ricotta, D.; Licenziati, S.; Solis, A.A.; Ausenda, S.; De Francesco, M.; Garrafa, E.; Simonini, A.; Imberti, L.; et al. Generation of CD28− cells from long-term-stimulated CD8+CD28+ T cells: A possible mechanism accounting for the increased number of CD8+CD28− T cells in HIV-1-infected patients. J. Leukoc. Biol. 1999, 65, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Maly, K.; Schirmer, M. The story of CD4+ CD28− T cells revisited: Solved or still ongoing? J. Immunol. Res. 2015, 2015, 348746. [Google Scholar] [PubMed] [Green Version]
- Mou, D.; Espinosa, J.; Lo, D.J.; Kirk, A.D. CD28 negative T cells: Is their loss our gain? Am. J. Transplant. 2014, 14, 2460–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thewissen, M.; Somers, V.; Hellings, N.; Fraussen, J.; Damoiseaux, J.; Stinissen, P. CD4+CD28null T cells in autoimmune disease: Pathogenic features and decreased susceptibility to immunoregulation. J. Immunol. 2007, 179, 6514–6523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, D.; Espinosa, J.E.; Stempora, L.; Iwakoshi, N.N.; Kirk, A.D. Viral-induced CD28 loss evokes costimulation independent alloimmunity. J. Surg. Res. 2015, 196, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett-Racke, A.E.; Trotter, J.L.; Lauber, J.; Perrin, P.J.; June, C.H.; Racke, M.K. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J. Clin. Investig. 1998, 101, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Markovic-Plese, S.; Cortese, I.; Wandinger, K.P.; McFarland, H.F.; Martin, R. CD4+CD28− costimulation-independent T cells in multiple sclerosis. J. Clin. Investig. 2001, 108, 1185–1194. [Google Scholar] [CrossRef]
- Broux, B.; Mizee, M.R.; Vanheusden, M.; Van Der Pol, S.; Van Horssen, J.; Van Wijmeersch, B.; Somers, V.; De Vries, H.E.; Stinissen, P.; Hellings, N. IL-15 Amplifies the Pathogenic Properties of CD4+CD28− T Cells in Multiple Sclerosis. J. Immunol. 2015, 194, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Broux, B.; Pannemans, K.; Zhang, X.; Markovic-Plese, S.; Broekmans, T.; Eijnde, B.O.; Van Wijmeersch, B.; Somers, V.; Geusens, P.; van der Pol, S.; et al. CX3CR1 drives cytotoxic CD4+CD28− T cells into the brain of multiple sclerosis patients. J. Autoimmun. 2012, 38, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Broux, B.; Markovic-Plese, S.; Stinissen, P.; Hellings, N. Pathogenic features of CD4+CD28− T cells in immune disorders. Trends Mol. Med. 2012, 18, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Hildemann, S.K.; Eberlein, J.; Davenport, B.; Nguyen, T.T.; Victorino, F.; Homann, D. High Efficiency of Antiviral CD4+ Killer T Cells. PLoS ONE 2013, 8, e60420. [Google Scholar]
- Vanheusden, M.; Broux, B.; Welten, S.P.; Peeters, L.M.; Panagioti, E.; Van Wijmeersch, B.; Somers, V.; Stinissen, P.; Arens, R.; Hellings, N. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci. Rep. 2017, 7, 663. [Google Scholar] [CrossRef]
- Peeters, L.M.; Vanheusden, M.; Somers, V.; Van Wijmeersch, B.; Stinissen, P.; Broux, B.; Hellings, N. Cytotoxic CD4+ T Cells Drive Multiple Sclerosis Progression. Front. Immunol. 2017, 8, 1160. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med. 2011, 3, 111ra20. [Google Scholar] [CrossRef] [Green Version]
- Asseman, C.; Mauze, S.; Leach, M.W.; Coffman, R.L.; Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999, 190, 995–1004. [Google Scholar] [CrossRef]
- Cuende, J.; Lienart, S.; Dedobbeleer, O.; van der Woning, B.; De Boeck, G.; Stockis, J.; Huygens, C.; Colau, D.; Somja, J.; Delvenne, P.; et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci. Transl. Med. 2015, 7, 284ra56. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Anderton, S.M. Cytokines in the induction and resolution of experimental autoimmune encephalomyelitis. Cytokine 2005, 32, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Strober, W. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 2001, 194, 629–644. [Google Scholar] [CrossRef]
- Strauss, L.; Bergmann, C.; Whiteside, T.L. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J. Immunol. 2009, 182, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Sitkovsky, M. Extracellular adenosine-mediated modulation of regulatory T cells. Front. Immunol. 2014, 5, 304. [Google Scholar] [CrossRef] [PubMed]
- Borsellino, G.; Kleinewietfeld, M.; Di Mitri, D.; Sternjak, A.; Diamantini, A.; Giometto, R.; Höpner, S.; Centonze, D.; Bernardi, G.; Dell’Acqua, M.L.; et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood 2007, 110, 1225–1232. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M.; et al. 70 Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [Green Version]
- Sumida, T.; Lincoln, M.R.; Ukeje, C.M.; Rodriguez, D.M.; Akazawa, H.; Noda, T.; Naito, A.T.; Komuro, I.; Dominguez-Villar, M.; Hafler, D.A. Activated beta-catenin in Foxp3+ regulatory T cells links inflammatory environments to autoimmunity. Nat. Immunol. 2018, 19, 1391–1402. [Google Scholar] [CrossRef]
- Bhela, S.; Kempsell, C.; Manohar, M.; Dominguez-Villar, M.; Griffin, R.; Bhatt, P.; Kivisakk-Webb, P.; Fuhlbrigge, R.; Kupper, T.; Weiner, H.; et al. Nonapoptotic and extracellular activity of granzyme B mediates resistance to regulatory T cell (Treg) suppression by HLA-DR-CD25hiCD127lo Tregs in multiple sclerosis and in response to IL-6. J. Immunol. 2015, 194, 2180–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cava, A. Tregs are regulated by cytokines: Implications for autoimmunity. Autoimmun. Rev. 2008, 8, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, B.J.; Thomas, H.E.; Pai, S.; Santamaria, P.; Iwakura, Y.; Steptoe, R.J.; Kay, T.W.; Thomas, R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J. Immunol. 2006, 176, 7278–7287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonderegger, I.; Iezzi, G.; Maier, R.; Schmitz, N.; Kurrer, M.; Kopf, M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J. Exp. Med. 2008, 205, 2281–2294. [Google Scholar] [CrossRef]
- Trinschek, B.; Lüssi, F.; Haas, J.; Wildemann, B.; Zipp, F.; Wiendl, H.; Becker, C.; Jonuleit, H. Kinetics of IL-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS ONE 2013, 8, e77634. [Google Scholar] [CrossRef]
- Albrecht, I.; Niesner, U.; Janke, M.; Menning, A.; Loddenkemper, C.; Kühl, A.A.; Lepenies, I.; Lexberg, M.H.; Westendorf, K.; Hradilkova, K.; et al. Persistence of effector memory Th1 cells is regulated by Hopx. Eur. J. Immunol. 2010, 40, 2993–3006. [Google Scholar] [CrossRef]
- Nocentini, G.; Riccardi, C. GITR: A multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur. J. Immunol. 2005, 35, 1016–1022. [Google Scholar] [CrossRef]
- Van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Jonuleit, H.; Schmitt, E.; Stassen, M.; Tuettenberg, A.; Knop, J.; Enk, A.H. Identification and functional characterization of human CD4+CD25+ T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 2001, 193, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Baatar, D.; Olkhanud, P.; Sumitomo, K.; Taub, D.; Gress, R.; Biragyn, A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4− Tregs, regulate effector T cells using FasL. J. Immunol. 2007, 178, 4891–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo, A.N.; Schirmer, M.; Weyand, C.M.; Goronzy, J.J. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J. Immunol. 2000, 165, 6301–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Cederbom, L.; Hall, H.; Ivars, F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 2000, 30, 1538–1543. [Google Scholar] [CrossRef]
- Pinto-Medel, M.J.; García-León, J.A.; Oliver-Martos, B.; López-Gómez, C.; Luque, G.; Arnáiz-Urrutia, C.; Órpez, T.; Marín-Bañasco, C.; Fernández, Ó.; Leyva, L. The CD4+ T-cell subset lacking expression of the CD28 costimulatory molecule is expanded and shows a higher activation state in multiple sclerosis. J. Neuroimmunol. 2012, 243, 1–11. [Google Scholar] [CrossRef]
- Van Bergen, J.; Thompson, A.; van der Slik, A.; Ottenhoff, T.H.; Gussekloo, J.; Koning, F. Phenotypic and functional characterization of CD4 T cells expressing killer Ig-like receptors. J. Immunol. 2004, 173, 6719–6726. [Google Scholar] [CrossRef]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef]
- Zielinski, C.E. Autoimmunity beyond Th17: GM-CSF producing T cells. Cell Cycle 2014, 13, 2489–2490. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W. IL-1 in Th17 Differentiation and Inflammatory Tissue Damage. Austin J. Clin. Immunol. 2014, 1, 4. [Google Scholar]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.M.; Branigan, P.J.; Bamezai, A. Interferon gamma enhances clonal expansion and survival of CD4+ T cells. J. Interferon Cytokine Res. 2008, 28, 611–622. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; McHugh, R.S.; Piccirillo, C.A.; Thornton, A.M. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol. Rev. 2001, 182, 58–67. [Google Scholar] [CrossRef]
- Abbas, A.K.; Trotta, E.; Simeonov, D.R.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appay, V.; Zaunders, J.J.; Papagno, L.; Sutton, J.; Jaramillo, A.; Waters, A.; Easterbrook, P.; Grey, P.; Smith, D.; McMichael, A.J.; et al. Characterization of CD4+ CTLs Ex Vivo. J. Immunol. 2002, 168, 5954–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, M.; Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28− T cells from rheumatoid arthritis patients. J. Immunol. 1998, 161, 1018–1025. [Google Scholar]
- Dominguez-Villar, M.; Baecher-Allan, C.M.; Hafler, D.A. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 2011, 17, 673–675. [Google Scholar] [CrossRef] [Green Version]
- Ihantola, E.L.; Viisanen, T.; Gazali, A.M.; Näntö-Salonen, K.; Juutilainen, A.; Moilanen, L.; Rintamäki, R.; Pihlajamäki, J.; Veijola, R.; Toppari, J.; et al. Effector T Cell Resistance to Suppression and STAT3 Signaling during the Development of Human Type 1 Diabetes. J. Immunol. 2018, 201, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitriu, I.E. The life (and death) of CD4+CD28null T cells in inflammatory diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, A.R.; Shin, E.C. Cytomegalovirus Infection and Memory T Cell Inflation. Immune Netw. 2015, 15, 186–190. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Full Name | Forward Primer | Reverse Primer |
|---|---|---|---|
| A2AR | Adenosine A2A receptor | CGCTCCGGTACAATGGCTT | TTGTTCCAACCTAGCATGGGA |
| CD39 | Ectonucleoside triphosphate diphosphohydrolase-1 | ACTATCGAGTCCCCAGATAATGC | CCTGATCCTTCCCATAGCACAA |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 | TGGATTTCAGCGGCACAAGGCT | CTGGGCCACGTGCATTGCTTTG |
| CYCA | Cyclin A | AGACTGAGTGGTTGGATGGC | TCGAGTTGTCCACAGTCAGC |
| FasL | Fas ligand | AAAGTGGCCCATTTAACAGGC | AAAGCAGGACAATTCCATAGGTG |
| Foxp3 | Forkhead box P3 | GCACCAGCTCTCAACGGTGGATG | GAAGACCCCAGTGGCGGTGG |
| GARP | Glycoprotein A Repetitions Predominant | AGACCCTTGATCTATCTGGGAAC | GAAGCTGATCTCATTGGTGCT |
| GITR | glucocorticoid-induced TNFR-related protein | CGAGTGGGACTGCATGTGTG | GGCAGGTCGTGCAGCAA |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor | CCAGGAGCCGACCTGCCTACA | GAAGTTTCCGGGGTTGGAGGGC |
| GrB | Granzyme B | GCGAATCTGACTTACGCCATTA | CCAGAGTCCCCCTTAAAGGAA |
| HOPX | Homeodomain-only protein | TCAACAAGGTCGACAAGCAC | TCTGTGACGGATCTGCACTC |
| IFN-γ | Interferon-γ | GGGGCCAACTAGGCAGCCAAC | AAGCACTGGCTCAGATTGCAGGC |
| IL-1β | Interleukin-1β | GATGAAGTGCTCCTTCCAGG | GCATCTTCCTCAGCTTGTCC |
| IL-6 | Interleukin-6 | GAGGAGACTTGCCTGGTGAA | GCTCTGGCTTGTTCCTCACT |
| IL-10 | Interleukin-10 | GCTGTCATCGATTTCTTCCC | ATAGAGTCGCCACCCTGATG |
| IL-10Rα | Interleukin-10Rα | CCTCCGTCTGTGTGGTTTGAA | CACTGCGGTAAGGTCATAGGA |
| IL-17 | Interleukin-17 | ATGGCCCAGCCATGGTCAAGTA | GCACAGGCGGGCAACTCTCA |
| IL-22 | Interleukin-22 | AACCGCACCTTCATGCTGGCT | CGCTCACTCATACTGACTCCGTGG |
| LAG3 | Lymphocyte-activation gene 3 | GCCTCCGACTGGGTCATTTT | CTTTCCGCTAAGTGGTGATGG |
| PD-1 | Programmed cell death protein 1 | CTCAGGGTGACAGAGAGAAG | GACACCAACCACCAGGGTTT |
| RPL13A | Ribosomal Protein L13a | AAGTTGAAGTACCTGGCTTTCC | GCCGTCAAACACCTTGAGAC |
| TGFβ | Transforming growth factor β | GTGGAAACCCACAACGAAAT | CACGTGCTGCTCCACTTTTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoeks, C.; Vanheusden, M.; Peeters, L.M.; Stinissen, P.; Broux, B.; Hellings, N. Treg-Resistant Cytotoxic CD4+ T Cells Dictate T Helper Cells in Their Vicinity: TH17 Skewing and Modulation of Proliferation. Int. J. Mol. Sci. 2021, 22, 5660. https://doi.org/10.3390/ijms22115660
Hoeks C, Vanheusden M, Peeters LM, Stinissen P, Broux B, Hellings N. Treg-Resistant Cytotoxic CD4+ T Cells Dictate T Helper Cells in Their Vicinity: TH17 Skewing and Modulation of Proliferation. International Journal of Molecular Sciences. 2021; 22(11):5660. https://doi.org/10.3390/ijms22115660
Chicago/Turabian StyleHoeks, Cindy, Marjan Vanheusden, Liesbet M. Peeters, Piet Stinissen, Bieke Broux, and Niels Hellings. 2021. "Treg-Resistant Cytotoxic CD4+ T Cells Dictate T Helper Cells in Their Vicinity: TH17 Skewing and Modulation of Proliferation" International Journal of Molecular Sciences 22, no. 11: 5660. https://doi.org/10.3390/ijms22115660
APA StyleHoeks, C., Vanheusden, M., Peeters, L. M., Stinissen, P., Broux, B., & Hellings, N. (2021). Treg-Resistant Cytotoxic CD4+ T Cells Dictate T Helper Cells in Their Vicinity: TH17 Skewing and Modulation of Proliferation. International Journal of Molecular Sciences, 22(11), 5660. https://doi.org/10.3390/ijms22115660