
Cerebral Pericytes and Endothelial Cells Communicate through Inflammasome-Dependent Signals

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results
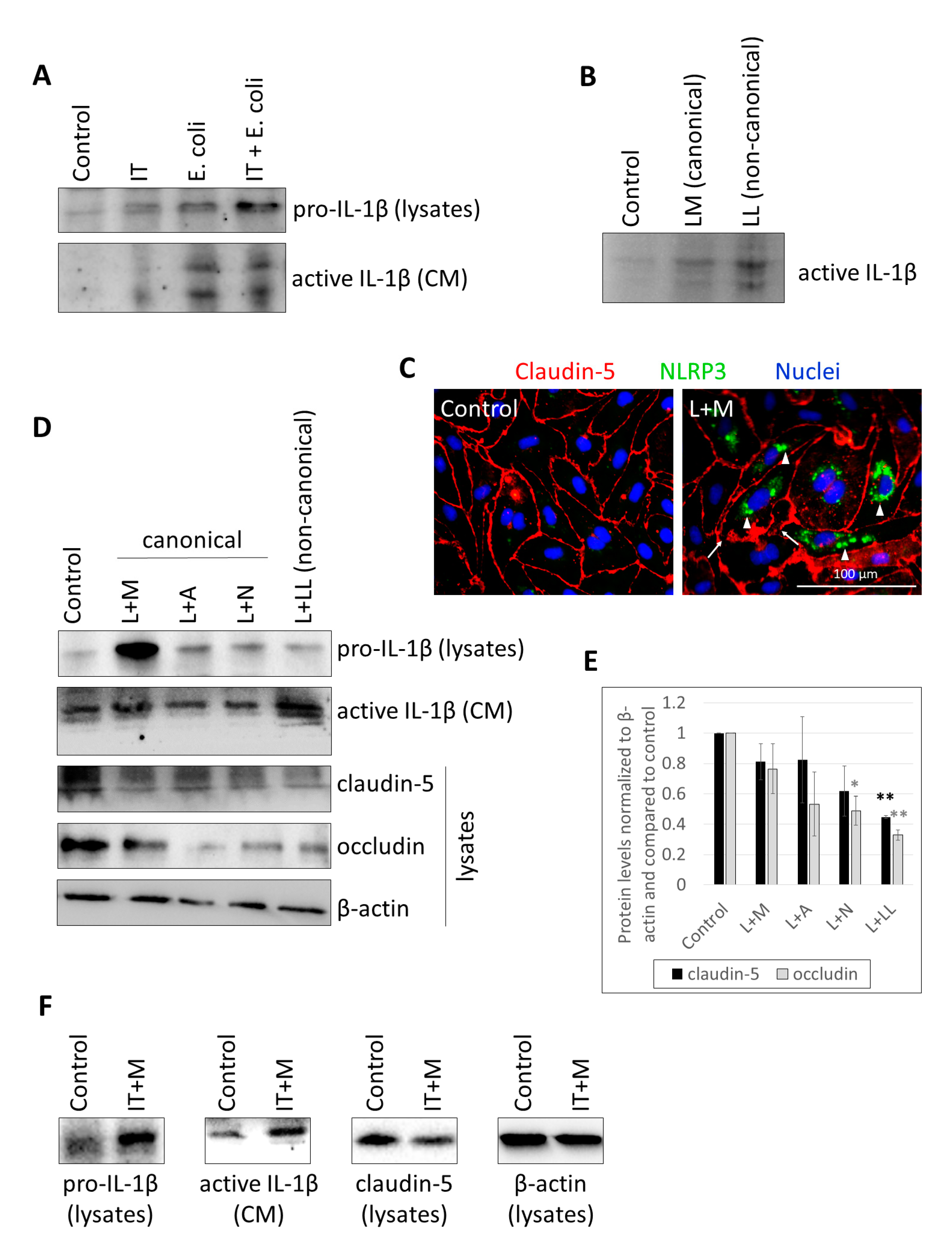
2.1. Characterization of Inflammasome Activation in CECs
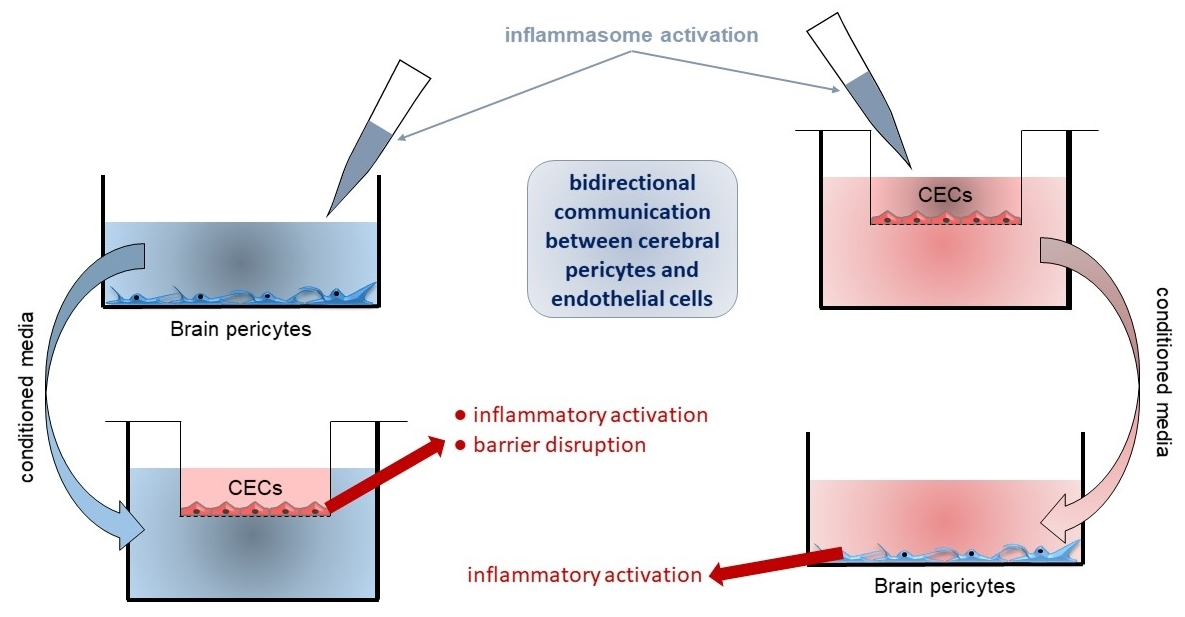
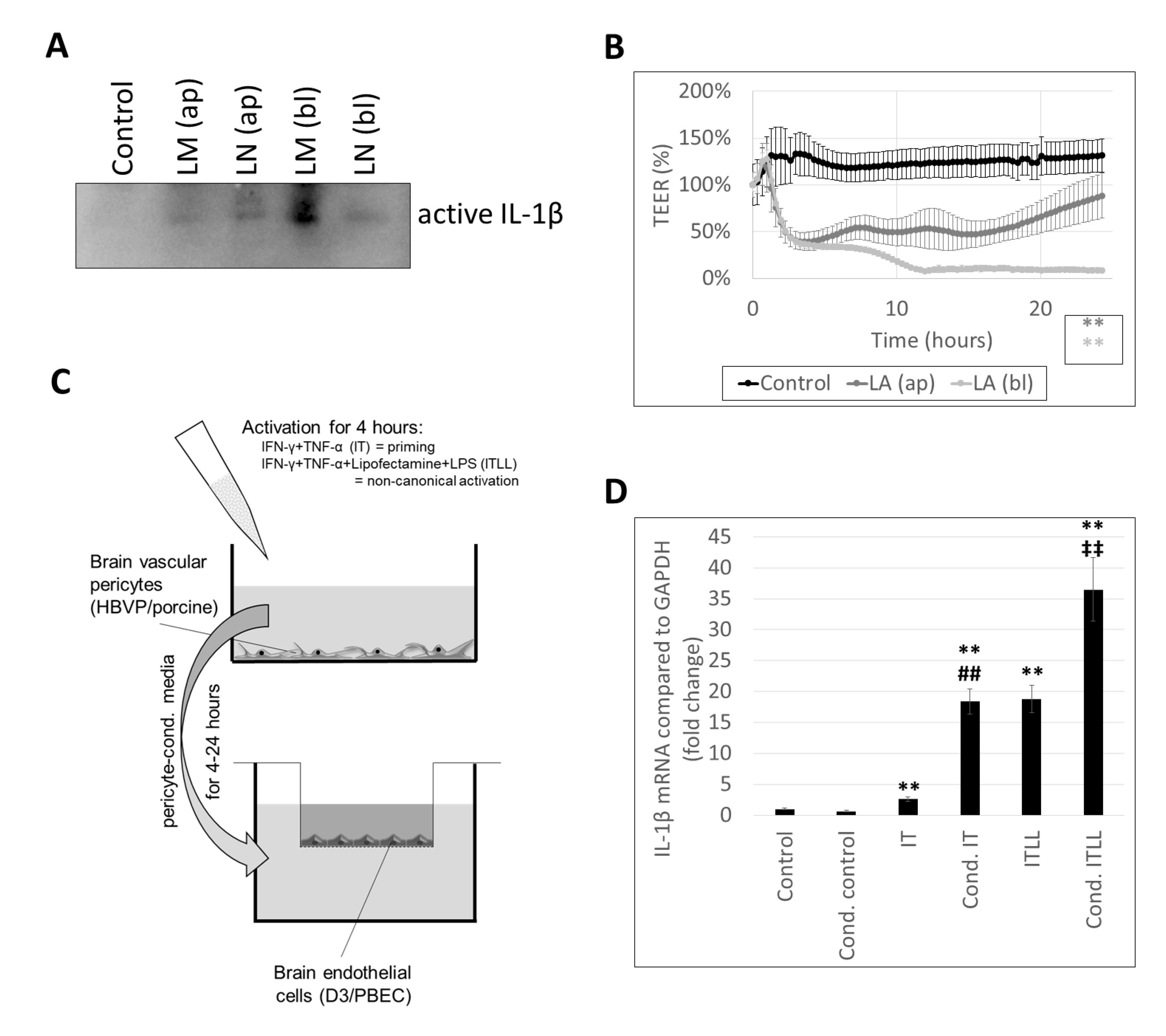
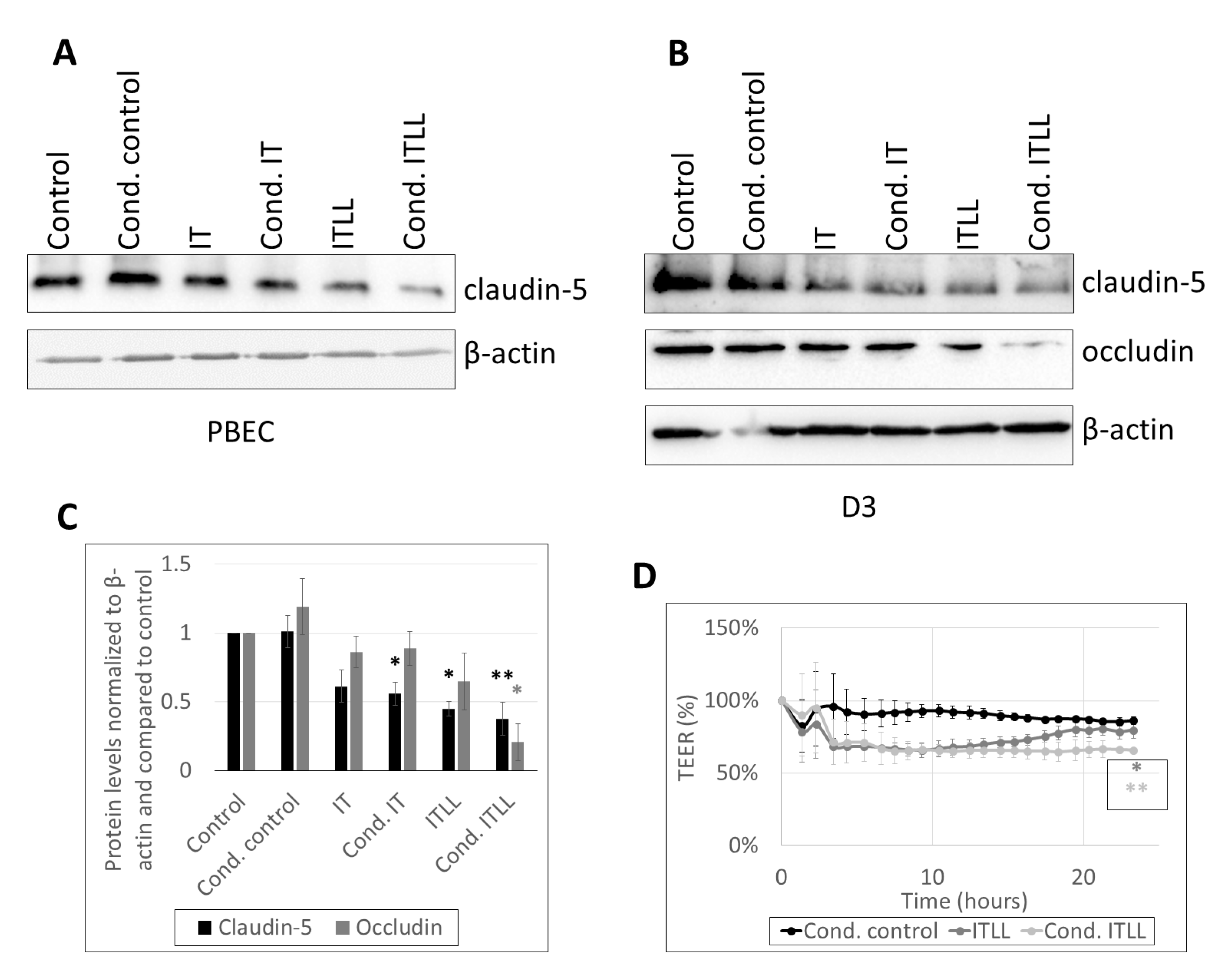
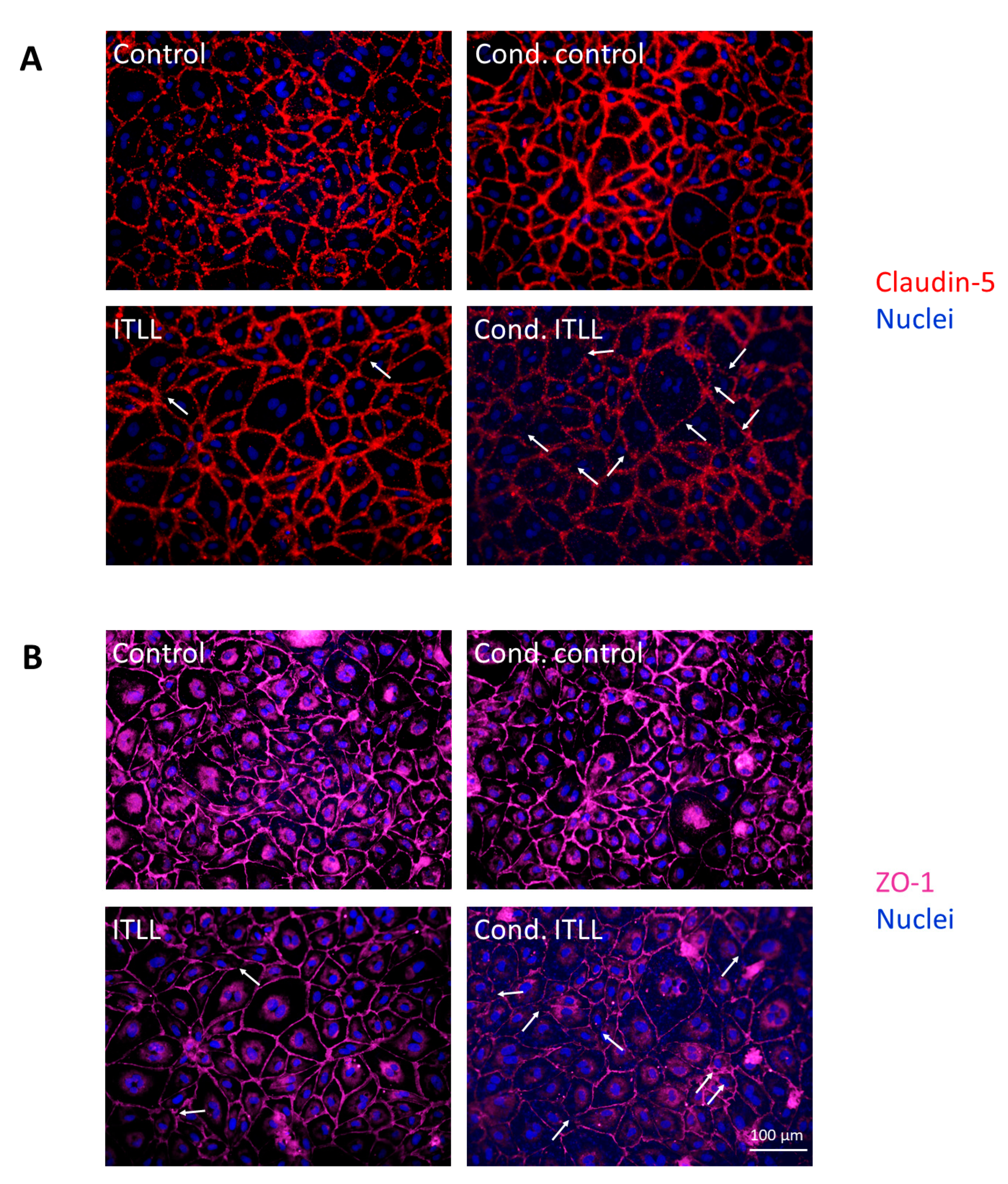
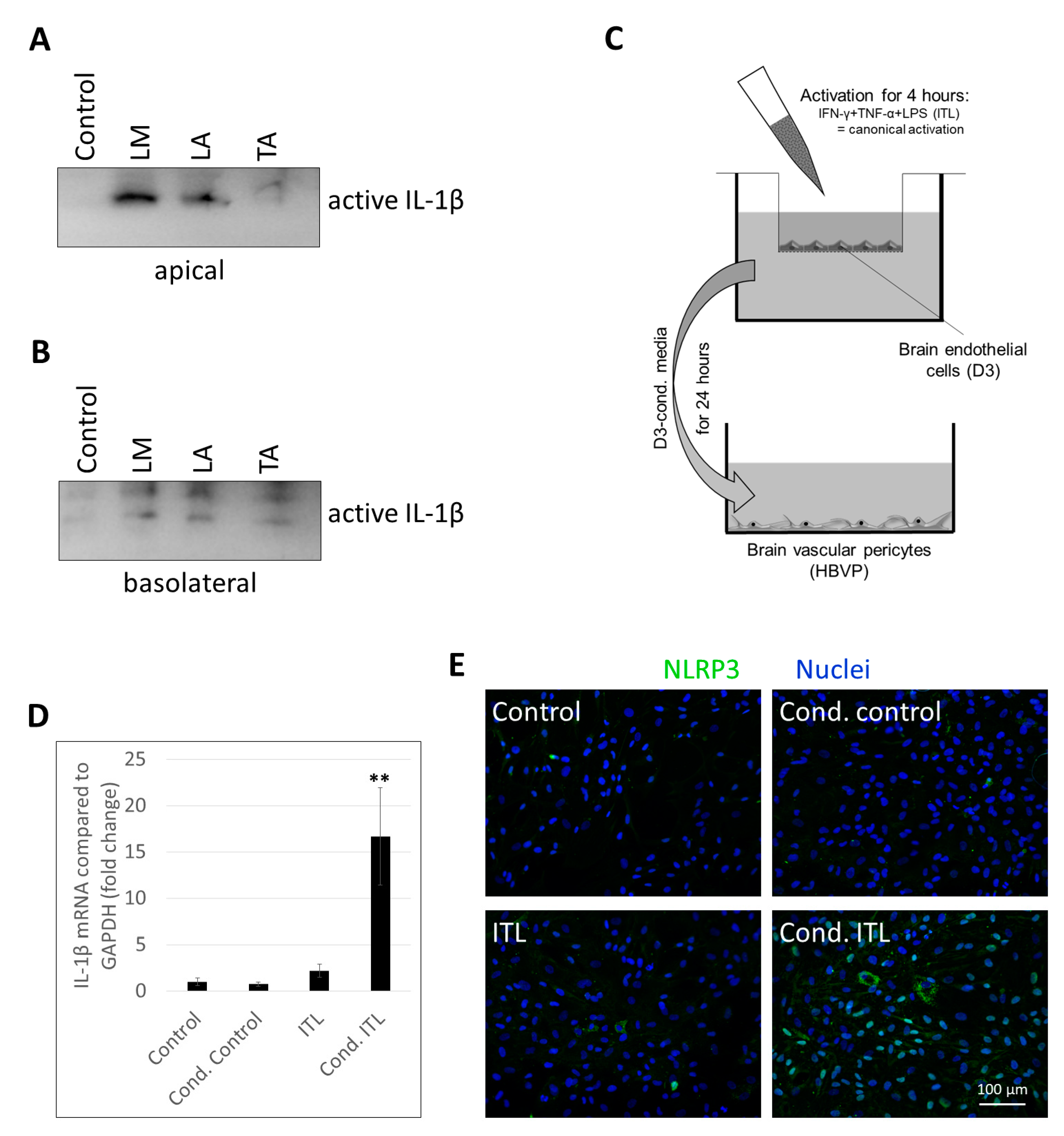
2.2. Activation of CECs by Pericytes Exposed to Inflammasome Activator Stimuli
2.3. Activation of Brain Pericytes by CECs Exposed to Inflammasome Activator Stimuli
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Brain Endothelial Cells and Pericytes
4.2. Inflammatory Activation of Brain Endothelial Cells and Pericytes
4.3. Coculture Systems for the Study of Endothelial–Pericyte Communication
4.4. Measurement of Transendothelial Electrical Resistance (TEER)
4.5. Sample Preparation and Western Blot
4.6. Immunofluorescence and Fluorescence Microscopy
4.7. Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mészáros, Á.; Molnár, K.; Nógrádi, B.; Hernádi, Z.; Nyúl-Tóth, Á.; Wilhelm, I.; Krizbai, I.A. Neurovascular inflammaging in health and disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163, 144–171. [Google Scholar] [CrossRef]
- Le Guennec, L.; Coureuil, M.; Nassif, X.; Bourdoulous, S. Strategies used by bacterial pathogens to cross the blood-brain barrier. Cell. Microbiol. 2019, 22, e13132. [Google Scholar] [CrossRef] [Green Version]
- Rochfort, K.D.; Cummins, P.M. The blood-brain barrier endothelium: A target for pro-inflammatory cytokines. Biochem. Soc. Trans. 2015, 43, 702–706. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costea, L.; Mészáros, Á.; Bauer, H.; Bauer, H.-C.; Traweger, A.; Wilhelm, I.; Farkas, A.E.; Krizbai, I.A. The blood-brain barrier and its intercellular junctions in age-related brain disorders. Int. J. Mol. Sci. 2019, 20, 5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. (War.) 2011, 71, 113–128. [Google Scholar]
- Bauer, H.C.; Traweger, A.; Zweimueller-Mayer, J.; Lehner, C.; Tempfer, H.; Krizbai, I.; Wilhelm, I. New aspects of the molecular constituents of tissue barriers. J. Neural Transm. 2010, 118, 7–21. [Google Scholar] [CrossRef]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Nakaoke, R.; Dohgu, S.; Banks, W.A. Release of cytokines by brain endothelial cells: A polarized response to lipopolysaccharide. Brain Behav. Immun. 2006, 20, 449–455. [Google Scholar] [CrossRef]
- Guijarro-Munoz, I.; Compte, M.; Álvarez-Cienfuegos, A.; Álvarez-Vallina, L.; Sanz, L. Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-kappaB signaling pathway and proin-flammatory response in human pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, J.; Takata, F.; Machida, T.; Takahashi, H.; Soejima, Y.; Funakoshi, M.; Futagami, K.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci. Lett. 2014, 578, 133–138. [Google Scholar] [CrossRef]
- Hurtado-Alvarado, G.; Cabañas-Morales, A.M.; Gómez-González, B. Pericytes: Brain-immune interface modulators. Front. Integr. Neurosci. 2014, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Nagyőszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.K.; Haskó, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef]
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Haskó, J.; Krizbai, I.A. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J. Neurochem. 2015, 135, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav. Immun. 2017, 64, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Stutz, A.; Horvath, G.L.; Monks, B.G.; Latz, E. ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol. 2013, 1040, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.-H.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 2018, 15, 1–18. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Schafer, D.P.; Vincent, A.; Blachère, N.E.; Bar-Or, A. Neuroinflammation: Ways in which the immune system affects the brain. Neurotherapeutics 2015, 12, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lénárt, N.; Brough, D.; Dénes, Á. Inflammasomes link vascular disease with neuroinflammation and brain disorders. Br. J. Pharmacol. 2016, 36, 1668–1685. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Nyúl-Tóth, Á.; Kozma, M.; Farkas, A.E.; Krizbai, I.A. Role of pattern recognition receptors of the neurovascular unit in inflammaging. Am. J. Physiol. Circ. Physiol. 2017, 313, H1000–H1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nógrádi, B.; Nyúl-Tóth, Á.; Kozma, M.; Molnár, K.; Patai, R.; Siklós, L.; Wilhelm, I.; Krizbai, I.A. Upregulation of nucleotide-binding oligomerization domain-, LRR- and pyrin domain-containing protein 3 in motoneurons following peripheral nerve injury in mice. Front. Pharmacol. 2020, 11, 584184. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the blood-brain barriers and blood-brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Banks, W.; Kovac, A.; Morofuji, Y. Neurovascular unit crosstalk: Pericytes and astrocytes modify cytokine secretion patterns of brain endothelial cells. Br. J. Pharmacol. 2018, 38, 1104–1118. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity 2019, 50, 317–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Mudher, A. Alzheimer’s disease and type 2 diabetes: A critical assessment of the shared pathological traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, Y.M.; Borcherding, D.; Kanthasamy, A.; Kim, H.J.; Willette, A.A.; Jergens, A.; Allenspach, K.; Mochel, J.P. The gut-brain axis in neurodegenerative diseases and relevance of the canine model: A review. Front. Aging Neurosci. 2019, 11, 130. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Dohgu, S.; Banks, W.A. Brain pericytes increase the lipopolysaccharide-enhanced transcytosis of HIV-1 free virus across the in vitro blood-brain barrier: Evidence for cytokine-mediated pericyte-endothelial cell crosstalk. Fluids Barriers CNS 2013, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyúl-Tóth, Á.; Suciu, M.; Molnár, J.; Fazakas, C.; Haskó, J.; Herman, H.; Farkas, A.E.; Kaszaki, J.; Hermenean, A.; Wilhelm, I.; et al. Differences in the molecular structure of the blood-brain barrier in the cerebral cortex and white matter: An in silico, in vitro, and ex vivo study. Am. J. Physiol. Circ. Physiol. 2016, 310, H1702–H1714. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Cat. No. | Application |
|---|---|---|
| anti-IL-1β g. polyclonal | AF-401-NA (R&D Systems/Bio-Techne) | WB: 1:500 in 1% BSA in TBS-T |
| anti-claudin-5 m. monoclonal | 35-2500 (Thermo Fisher Scientific) | WB: 1:300 in TBS-T |
| IF: 1:50 in 1% BSA in PBS | ||
| anti-occludin g. polyclonal | sc-8145 (Santa Cruz Biotechnology) | WB: 1:500 in TBS-T |
| anti-β-actin m. monoclonal | AC-15 (Merck-Sigma) | WB: 1:1000 in 1% BSA in TBS-T |
| anti-ZO-1 r. polyclonal | 61-7300 (Thermo Fisher Scientific) | IF: 1:50 in 1% BSA in PBS |
| anti-NLRP3 g. polyclonal | GTX88190 (GeneTex, Hsinchu City, Taiwan) | IF: 1:200 in 1% BSA in PBS |
| Secondary Antibody | Cat. No. | Application |
| HRP-conjugated g. anti-mouse IgG (H + L) | 610094 (BD Biosciences, San Jose, CA, USA) | WB: 1:3000 in TBS-T |
| HRP-conjugated g. anti-rabbit IgG (H + L) | 7074 (Cell Signaling Technology) | WB: 1:4000 in TBS-T |
| Alexa Fluor® 594 AffiniPure g. anti-mouse IgG (H + L) | 115-585-003 (Jackson Immuno Research, Ely, UK) | IF: 1:600 in 1% BSA in PBS |
| Alexa Fluor® 647 AffiniPure g. anti-rabbit IgG (H + L) | 111-605-003 (Jackson Immuno Research) | IF: 1:600 in 1% BSA in PBS |
| Alexa Fluor® 488 Cross-Adsorbed d. anti-goat IgG (H + L) | A-11055 (Thermo Fisher Scientific) | IF: 1:600 in 1% BSA in PBS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozma, M.; Mészáros, Á.; Nyúl-Tóth, Á.; Molnár, K.; Costea, L.; Hernádi, Z.; Fazakas, C.; Farkas, A.E.; Wilhelm, I.; Krizbai, I.A. Cerebral Pericytes and Endothelial Cells Communicate through Inflammasome-Dependent Signals. Int. J. Mol. Sci. 2021, 22, 6122. https://doi.org/10.3390/ijms22116122
Kozma M, Mészáros Á, Nyúl-Tóth Á, Molnár K, Costea L, Hernádi Z, Fazakas C, Farkas AE, Wilhelm I, Krizbai IA. Cerebral Pericytes and Endothelial Cells Communicate through Inflammasome-Dependent Signals. International Journal of Molecular Sciences. 2021; 22(11):6122. https://doi.org/10.3390/ijms22116122
Chicago/Turabian StyleKozma, Mihály, Ádám Mészáros, Ádám Nyúl-Tóth, Kinga Molnár, Laura Costea, Zsófia Hernádi, Csilla Fazakas, Attila E. Farkas, Imola Wilhelm, and István A. Krizbai. 2021. "Cerebral Pericytes and Endothelial Cells Communicate through Inflammasome-Dependent Signals" International Journal of Molecular Sciences 22, no. 11: 6122. https://doi.org/10.3390/ijms22116122
APA StyleKozma, M., Mészáros, Á., Nyúl-Tóth, Á., Molnár, K., Costea, L., Hernádi, Z., Fazakas, C., Farkas, A. E., Wilhelm, I., & Krizbai, I. A. (2021). Cerebral Pericytes and Endothelial Cells Communicate through Inflammasome-Dependent Signals. International Journal of Molecular Sciences, 22(11), 6122. https://doi.org/10.3390/ijms22116122