
Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway

 , , ,
, , ,  , , and
, , and
Abstract
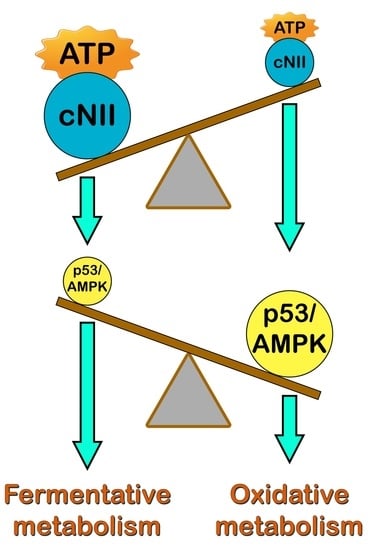
:
1. Introduction
2. Results
2.1. cN-II Silencing Affects Nucleotide Content in NCI-H292 Cells
2.2. Effect of cN-II Silencing on Proliferation of NCI-H292 Cells
2.3. cN-II Silencing Increases Oxidative Metabolism and Thiol Content in NCI-H292 Cells
2.4. The AMPK/mTOR Signaling Pathway Is Affected by cN-II Silencing in NCI-H292 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Cell Proliferation Assay
4.3. High Performance Capillary Electrophoresis (HPCE) Analysis
4.4. Quantification of Reduced Thiols
4.5. Enzymatic Assays
4.6. Western Blotting
4.7. Lactate Concentration Measurement
4.8. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, J.; Wang, J.; Chen, C.; Yuan, H.; Wen, X.; Sun, H. USP7: Target Validation and Drug Discovery for Cancer Therapy. Med. Chem. 2018, 14, 3–18. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Dogan, F.; Biray Avci, C. Correlation between telomerase and mTOR pathway in cancer stem cells. Gene 2018, 641, 235–239. [Google Scholar] [CrossRef]
- Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Hughes-Hallett, J.; Timson, R.C.; Ilagan, E.; Yuan, M.; Asara, J.M.; Ben-Sahra, I.; Manning, B.D. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Wallden, K.; Stenmark, P.; Nyman, T.; Flodin, S.; Graslund, S.; Loppnau, P.; Bianchi, V.; Nordlund, P. Crystal structure of human cytosolic 5’-nucleotidase II: Insights into allosteric regulation and substrate recognition. J. Biol. Chem. 2007, 282, 17828–17836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesi, R.; Petrotto, E.; Colombaioni, L.; Allegrini, S.; Garcia-Gil, M.; Camici, M.; Jordheim, L.P.; Tozzi, M.G. Cytosolic 5’-Nucleotidase II Silencing in a Human Lung Carcinoma Cell Line Opposes Cancer Phenotype with a Concomitant Increase in p53 Phosphorylation. Int. J. Mol. Sci. 2018, 19, 2115. [Google Scholar] [CrossRef] [Green Version]
- Pesi, R.; Allegrini, S.; Balestri, F.; Garcia-Gil, M.; Cividini, F.; Colombaioni, L.; Jordheim, L.P.; Camici, M.; Tozzi, M.G. Cytosolic 5′-Nucleotidase II Is a Sensor of Energy Charge and Oxidative Stress: A Possible Function as Metabolic Regulator. Cells 2021, 10, 182. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Pesi, R.; Allegrini, S. On the physiological role of cytosolic 5′-nucleotidase II (cN-II): Pathological and therapeutical implications. Curr. Med. Chem. 2013, 20, 4285–4291. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L.; Tozzi, M.G. Recent advances in structure and function of cytosolic IMP-GMP specific 5′-nucleotidase II (cN-II). Purinergic Signal. 2006, 2, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Tozzi, M.G. Metabolic Aspects of Adenosine Functions in the Brain. Front. Pharmacol. 2021, 12, 672182. [Google Scholar] [CrossRef] [PubMed]
- Cividini, F.; Tozzi, M.G.; Galli, A.; Pesi, R.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Allegrini, S. Cytosolic 5′-Nucleotidase II Interacts with the Leucin Rich Repeat of NLR Family Member Ipaf. PLoS ONE 2015, 10, e0121525. [Google Scholar] [CrossRef] [Green Version]
- Cividini, F.; Cros-Perrial, E.; Pesi, R.; Machon, C.; Allegrini, S.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Tozzi, M.G. Cell proliferation and drug sensitivity of human glioblastoma cells are altered by the stable modulation of cytosolic 5′-nucleotidase II. Int. J. Biochem. Cell Biol. 2015, 65, 222–229. [Google Scholar] [CrossRef]
- Bricard, G.; Cadassou, O.; Cassagnes, L.E.; Cros-Perrial, E.; Payen-Gay, L.; Puy, J.Y.; Lefebvre-Tournier, I.; Tozzi, M.G.; Dumontet, C.; Jordheim, L.P. The cytosolic 5’-nucleotidase cN-II lowers the adaptability to glucose deprivation in human breast cancer cells. Oncotarget 2017, 8, 67380–67393. [Google Scholar] [CrossRef]
- Johanns, M.; Kviklyte, S.; Chuang, S.J.; Corbeels, K.; Jacobs, R.; Herinckx, G.; Vertommen, D.; Schakman, O.; Duparc, T.; Cani, P.D.; et al. Genetic deletion of soluble 5′-nucleotidase II reduces body weight gain and insulin resistance induced by a high-fat diet. Mol. Genet. Metab. 2019, 126, 377–387. [Google Scholar] [CrossRef]
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Mizusawa, S.; Teranishi, H.; Matsuo, T.; Nakata, Y.; Hyogo, H.; Ochi, H.; Nakamura, T.; et al. Genetic variations in the CYP17A1 and NT5C2 genes are associated with a reduction in visceral and subcutaneous fat areas in Japanese women. J. Hum. Genet. 2012, 57, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Camici, M.; Garcia-Gil, M.; Allegrini, S.; Pesi, R.; Tozzi, M.G. Evidence for a Cross-Talk Between Cytosolic 5′-Nucleotidases and AMP-Activated Protein Kinase. Front. Pharmacol. 2020, 11, 609849. [Google Scholar] [CrossRef]
- Duarte, R.R.R.; Bachtel, N.D.; Cotel, M.C.; Lee, S.H.; Selvackadunco, S.; Watson, I.A.; Hovsepian, G.A.; Troakes, C.; Breen, G.D.; Nixon, D.F.; et al. The Psychiatric Risk Gene NT5C2 Regulates Adenosine Monophosphate-Activated Protein Kinase Signaling and Protein Translation in Human Neural Progenitor Cells. Biol. Psychiatry 2019, 86, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Kviklyte, S.; Vertommen, D.; Yerna, X.; Andersen, H.; Xu, X.F.; Gailly, P.; Bohlooly-Y, M.; Oscarsson, J.; Rider, M.H. Effects of genetic deletion of soluble 5′-nucleotidases NT5C1A and NT5C2 on AMPK activation and nucleotide levels in contracting mouse skeletal muscles. Am. J. Physiol. Endoc. Metab. 2017, 313, E48–E62. [Google Scholar] [CrossRef] [PubMed]
- Bricard, G.; Cros-Perrial, E.; Machon, C.; Dumontet, C.; Jordheim, L.P. Stably transfected adherent cancer cell models with decreased expression of 5′-nucleotidase cN-II. Nucleosides Nucleotides Nucleic Acids 2016, 35, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [Green Version]
- Eymin, B.; Gazzeri, S. Role of cell cycle regulators in lung carcinogenesis. Cell Adh. Mig. 2010, 4, 114–123. [Google Scholar] [CrossRef]
- Wu, S.B.; Wu, Y.T.; Wu, T.P.; Wei, Y.H. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1331–1344. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Careddu, M.G.; Allegrini, S.; Pesi, R.; Camici, M.; Garcia-Gil, M.; Tozzi, M.G. Knockdown of cytosolic 5′-nucleotidase II (cN-II) reveals that its activity is essential for survival in astrocytoma cells. Biochim. Biophys. Acta-Mol. Cell Res. 2008, 1783, 1529–1535. [Google Scholar] [CrossRef] [Green Version]
- Cadassou, O.; Raza, M.Z.; Machon, C.; Gudefin, L.; Armanet, C.; Chettab, K.; Guitton, J.; Tozzi, M.G.; Dumontet, C.; Cros-Perrial, E.; et al. Enhanced migration of breast and lung cancer cells deficient for cN-II and CD73 via COX-2/PGE2/AKT axis regulation. Cell Oncol. 2020. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabutis, N.; Dimitropoulou, C.; Birmpas, C.; Joshi, A.; Thangjam, G.; Catravas, J.D. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L776–L787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Singh, M.; Selivanova, G.; Peuget, S. Pifithrin-alpha alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10, 1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.S.; Chiang, T.H.; Wang, J.S.; Lin, L.J.; Chao, W.C.; Inbaraj, B.S.; Lu, J.F.; Chen, B.H. Induction of p53-independent growth inhibition in lung carcinoma cell A549 by gypenosides. J Cell Mol Med 2015, 19, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Dastjerdi, M.N.; Rarani, M.Z.; Valiani, A.; Mahmoudieh, M. The effect of adenosine A1 receptor agonist and antagonist on p53 and caspase 3, 8, and 9 expression and apoptosis rate in MCF-7 breast cancer cell line. Res. Pharm. Sci. 2016, 11, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Jordheim, L.P.; Puy, J.Y.; Cros-Perrial, E.; Peyrottes, S.; Lefebvre, I.; Perigaud, C.; Dumontet, C. Determination of the enzymatic activity of cytosolic 5′-nucleotidase cN-II in cancer cells: Development of a simple analytical method and related cell line models. Anal. Bioanal. Chem. 2015, 407, 5747–5758. [Google Scholar] [CrossRef]
- Chiba, K.; Kawakami, K.; Tohyama, K. Simultaneous evaluation of cell viability by neutral red, MTT and crystal violet staining assays of the same cells. Toxicol. In Vitro 1998, 12, 251–258. [Google Scholar] [CrossRef]
- Allegrini, S.; Filoni, D.N.; Galli, A.; Collavoli, A.; Pesi, R.; Camici, M.; Tozzi, M.G. Expression of Bovine Cytosolic 5’-Nucleotidase (cN-II) in Yeast: Nucleotide Pools Disturbance and Its Consequences on Growth and Homologous Recombination. PLoS ONE 2013, 8, e63914. [Google Scholar] [CrossRef]
- Cividini, F.; Filoni, D.N.; Pesi, R.; Allegrini, S.; Camici, M.; Tozzi, M.G. IMP–GMP specific cytosolic 5′-nucleotidase regulates nucleotide pool and prodrug metabolism. Biochim. Biophys. Acta 2015, 1850, 1354–1361. [Google Scholar] [CrossRef]
- Peterson, G.L. Determination of total protein. Methods Enzymol. 1983, 91, 95–119. [Google Scholar] [CrossRef]
- Aykac, G.; Uysal, M.; Yalcin, A.S.; Kocaktoker, N.; Sivas, A.; Oz, H. The Effect of Chronic Ethanol Ingestion on Hepatic Lipid Peroxide, Glutathione, Glutathione-Peroxidase and Glutathione Transferase in Rats. Toxicology 1985, 36, 71–76. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gerber, G.; Preissler, H.; Heinrich, R.; Rapoport, S.M. Hexokinase of human erythrocytes. Purification, kinetic model and its application to the conditions in the cell. Eur. J. Biochem. 1974, 45, 39–52. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Camici, M.; Pesi, R.; Allegrini, S.; Sgarrella, F.; Ipata, P.L. Nucleoside phosphotransferase activity of human colon carcinoma cytosolic 5’-nucleotidase. Arch. Biochem. Biophys. 1991, 291, 212–217. [Google Scholar] [CrossRef]
- Liaud, N.; Navarro, D.; Vidal, N.; Sigoillot, J.C.; Raouche, S. High throughput automated colorimetric method for the screening of l-lactic acid producing microorganisms. MethodsX 2014, 1, 254–257. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCI-H292 Cell Line | with Pifithrin-α | without Pifithrin-α |
|---|---|---|
| pScont | 2.3 ± 0.1 | 2.0 ± 0.2 |
| pScNII | 0.7 ± 0.03 **** | 0.8 ± 0.01 **** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesi, R.; Allegrini, S.; Garcia-Gil, M.; Piazza, L.; Moschini, R.; Jordheim, L.P.; Camici, M.; Tozzi, M.G. Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 7004. https://doi.org/10.3390/ijms22137004
Pesi R, Allegrini S, Garcia-Gil M, Piazza L, Moschini R, Jordheim LP, Camici M, Tozzi MG. Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(13):7004. https://doi.org/10.3390/ijms22137004
Chicago/Turabian StylePesi, Rossana, Simone Allegrini, Mercedes Garcia-Gil, Lucia Piazza, Roberta Moschini, Lars Petter Jordheim, Marcella Camici, and Maria Grazia Tozzi. 2021. "Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway" International Journal of Molecular Sciences 22, no. 13: 7004. https://doi.org/10.3390/ijms22137004
APA StylePesi, R., Allegrini, S., Garcia-Gil, M., Piazza, L., Moschini, R., Jordheim, L. P., Camici, M., & Tozzi, M. G. (2021). Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway. International Journal of Molecular Sciences, 22(13), 7004. https://doi.org/10.3390/ijms22137004