
p53 Activation in Genetic Disorders: Different Routes to the Same Destination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
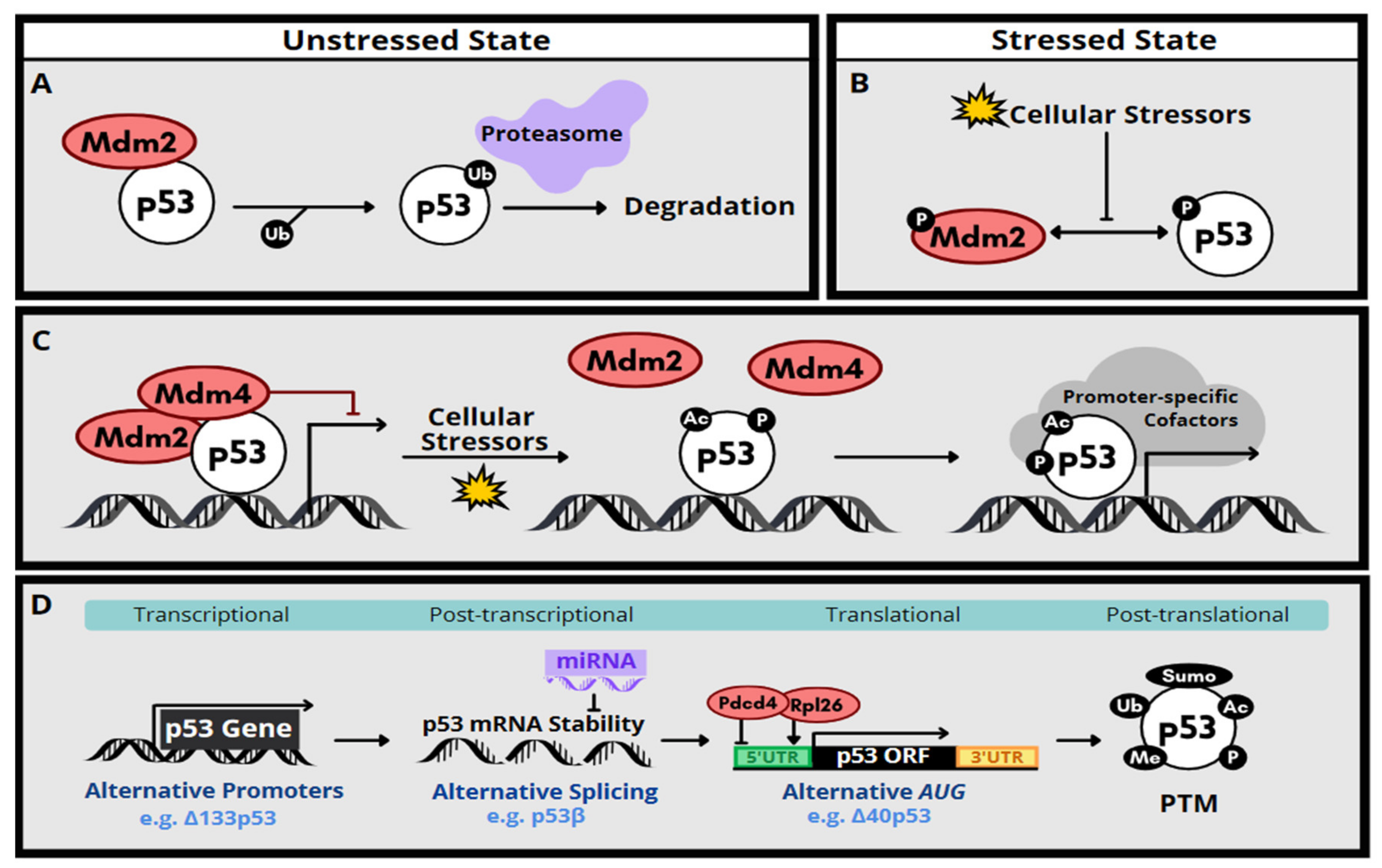
2. Molecular Function and Regulation of p53
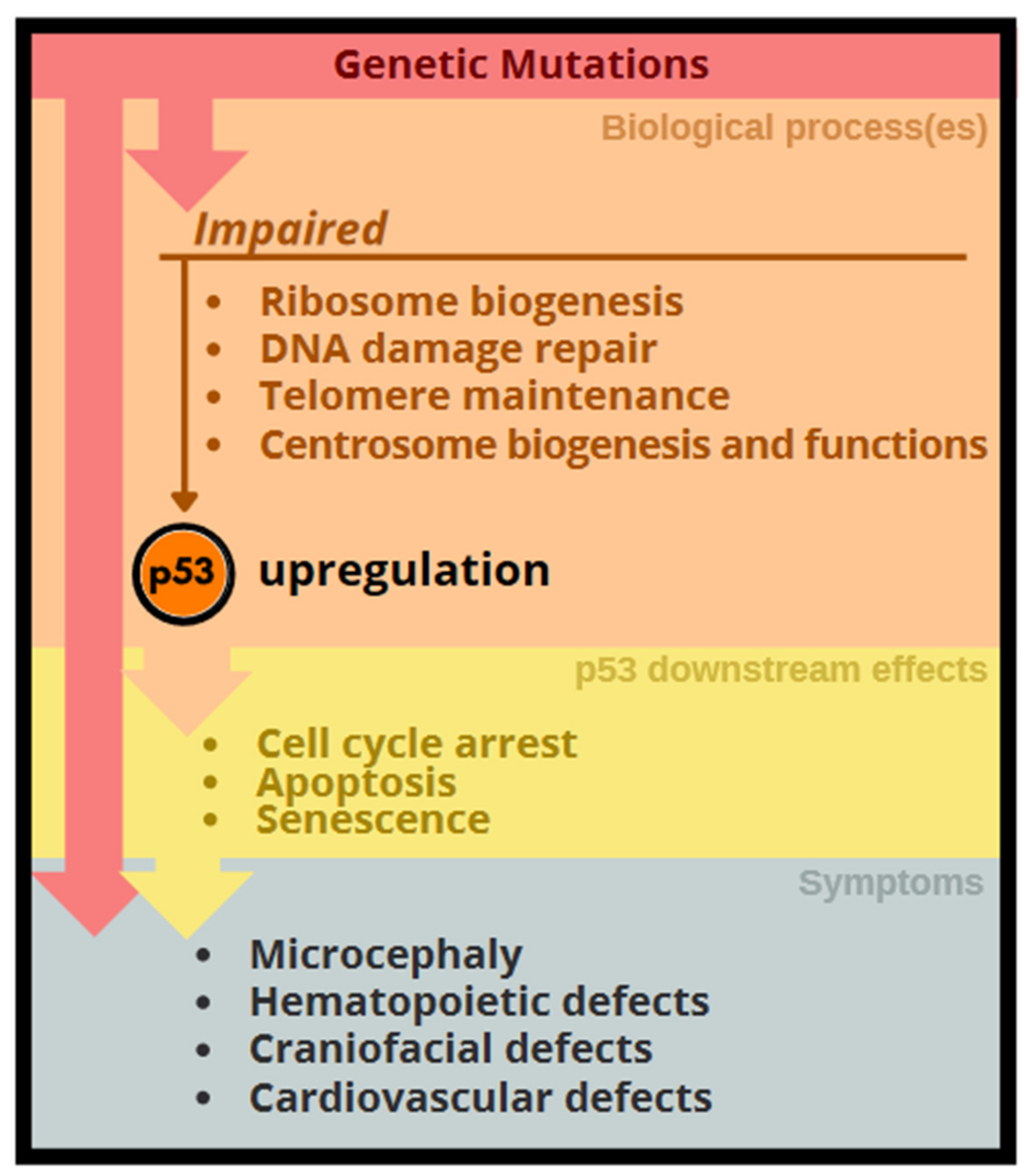
3. p53 Activation Associated with Congenital Anomalies
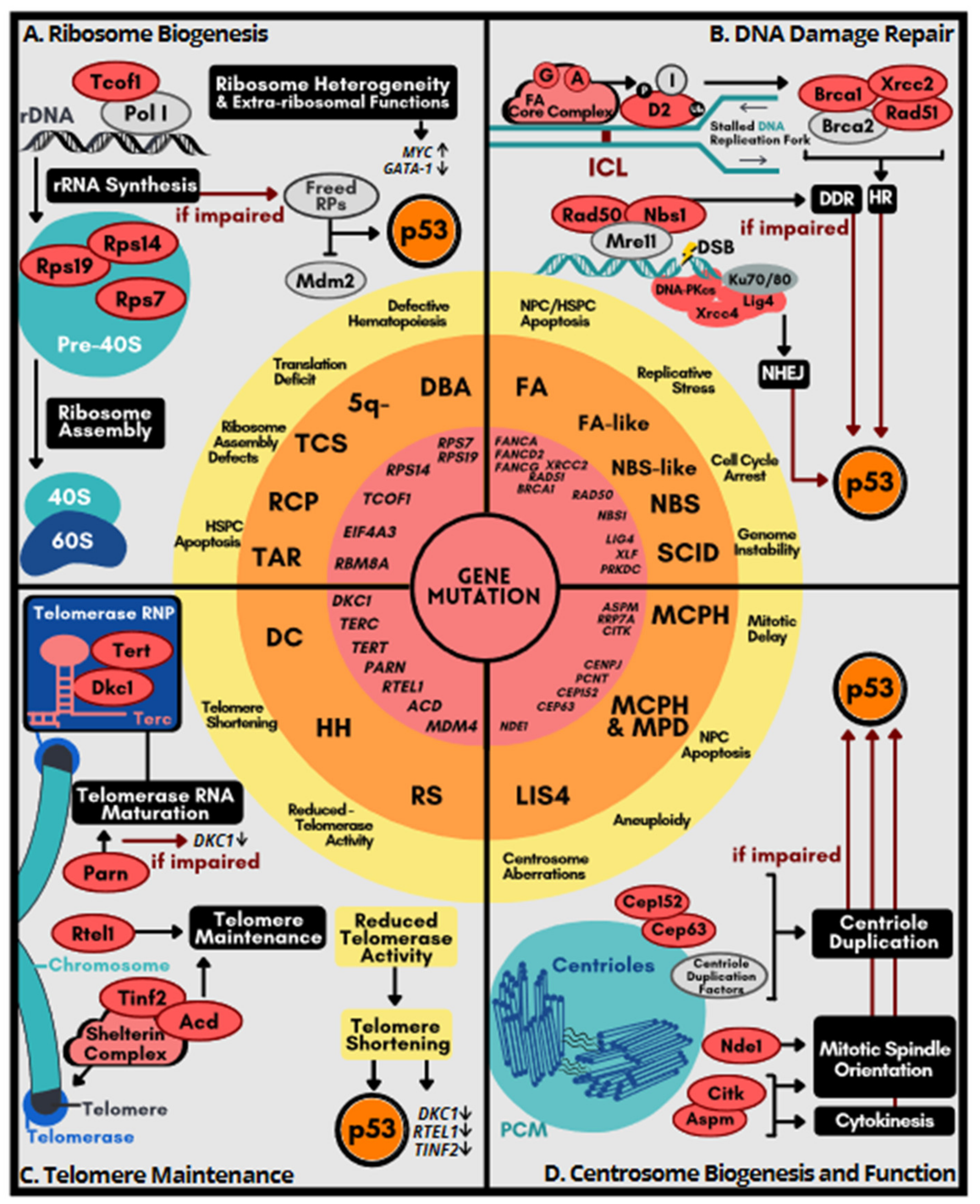
4. Genetic Disorders Related to Excessive p53 Activation
4.1. Disorders of Ribosome Dysfunction
4.2. Disorders Related to DNA Repair Deficiency
4.3. Syndromes Caused by Telomere Dysfunction
4.4. Disorders Related to Centrosome Dysfunction
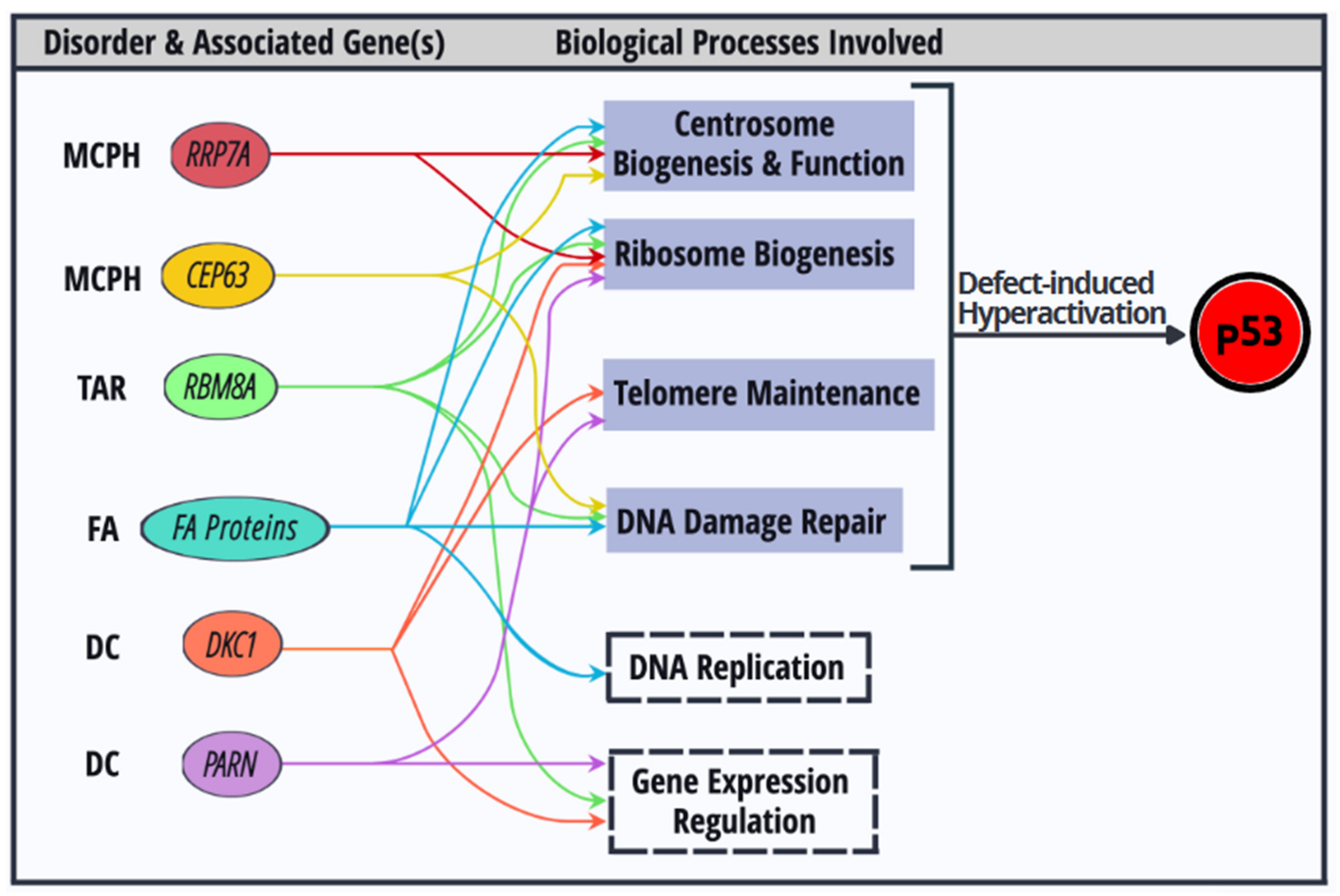
5. Interrelationships of the Cellular Processes Implicated in p53 Activation-Associated Disorders
6. Therapeutic Strategies for p53 Activation-Associated Disorders
7. Conclusions and Outlook
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Andrysik, Z.; Galbraith, M.D.; Guarnieri, A.L.; Zaccara, S.; Sullivan, K.D.; Pandey, A.; MacBeth, M.; Inga, A.; Espinosa, J.M. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017, 27, 1645–1657. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [Green Version]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Jain, A.K.; Barton, M.C. p53: Emerging roles in stem cells, development and beyond. Development 2018, 145, dev158360. [Google Scholar] [CrossRef] [Green Version]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szybińska, A.; Leśniak, W. P53 Dysfunction in Neurodegenerative Diseases - The Cause or Effect of Pathological Changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Bowen, M.E.; Attardi, L.D. The role of p53 in developmental syndromes. J. Mol. Cell Biol. 2019, 11, 200–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Lucchesi, C.; Zhang, J.; Chen, X. Modulation of the p53 family network by RNA-binding proteins. Transl. Cancer Res. 2016, 5, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutiérrez, N.C. Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63, correction in 2005, 123, 536–537. [Google Scholar] [CrossRef]
- Wedeken, L.; Singh, P.; Klempnauer, K.H. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J. Biol. Chem. 2011, 286, 42855–42862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, V.; Dichtel-Danjoy, M.L.; Sagne, C.; Hafsi, H.; Ma, D.; Ortiz-Cuaran, S.; Olivier, M.; Hall, J.; Mollereau, B.; Hainaut, P.; et al. Biological functions of p53 isoforms through evolution: Lessons from animal and cellular models. Cell Death Differ. 2011, 18, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fåhraeus, R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002, 4, 462–467, correction in 2002, 4, 912. [Google Scholar] [CrossRef] [PubMed]
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.C.; Lane, D.P.; Peng, J. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef] [Green Version]
- Bowen, M.E.; McClendon, J.; Long, H.K.; Sorayya, A.; Van Nostrand, J.L.; Wysocka, J.; Attardi, L.D. The Spatiotemporal Pattern and Intensity of p53 Activation Dictates Phenotypic Diversity in p53-Driven Developmental Syndromes. Dev. Cell 2019, 50, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Mendrysa, S.M.; O’Leary, K.A.; McElwee, M.K.; Michalowski, J.; Eisenman, R.N.; Powell, D.A.; Perry, M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006, 20, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.V.; Leblanc, M.; Fox, N.; Mao, J.H.; Tinkum, K.L.; Krummel, K.; Engle, D.; Piwnica-Worms, D.; Piwnica-Worms, H.; Balmain, A.; et al. Fine-tuning p53 activity through C-terminal modification significantly contributes to HSC homeostasis and mouse radiosensitivity. Genes Dev. 2011, 25, 1426–1438. [Google Scholar] [CrossRef] [Green Version]
- Van Nostrand, J.L.; Brady, C.A.; Jung, H.; Fuentes, D.R.; Kozak, M.M.; Johnson, T.M.; Lin, C.Y.; Lin, C.J.; Swiderski, D.L.; Vogel, H.; et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014, 514, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group. Nat Genet. 1996, 12, 130–136. [CrossRef] [PubMed]
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.P.; Dixon, M.J.; Trainor, P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.C.; Lynn, M.L.; Gaudenz, K.; Sakai, D.; Aoto, K.; Rey, J.P.; Glynn, E.F.; Ellington, L.; Du, C.; Dixon, J.; et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008, 14, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Franklin, D.A.; Zhang, Y. The Evolution of the Ribosomal Protein-MDM2-p53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026138. [Google Scholar] [CrossRef] [Green Version]
- Holohan, B.; Wright, W.E.; Shay, J.W. Cell biology of disease: Telomeropathies: An emerging spectrum disorder. J. Cell Biol. 2014, 205, 289–299. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, I.; Kim, J.; Kim, K. DNA damage to human genetic disorders with neurodevelopmental defects. J. Genet. Med. 2016, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, R.S.; Schoborg, T.A.; Rusan, N.M. Same but different: Pleiotropy in centrosome-related microcephaly. Mol. Biol. Cell 2018, 29, 241–246. [Google Scholar] [CrossRef]
- Van Nostrand, J.L.; Attardi, L.D. Guilty as CHARGED: p53’s expanding role in disease. Cell Cycle 2014, 13, 3798–3807. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.; Miriyala, K. Neural tube defects in Waardenburg syndrome: A case report and review of the literature. Am. J. Med. Genet. A 2017, 173, 2472–2477. [Google Scholar] [CrossRef]
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2020, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.S.; Freedman, M.H. Diamond-blackfan anemia: Etiology, pathophysiology, and treatment. Am. J. Pediatr. Hematol. Oncol. 1989, 11, 380–394. [Google Scholar]
- Jaako, P.; Flygare, J.; Olsson, K.; Quere, R.; Ehinger, M.; Henson, A.; Ellis, S.; Schambach, A.; Baum, C.; Richter, J.; et al. Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood 2011, 118, 6087–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 2010, 16, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Farley-Barnes, K.I.; Ogawa, L.M.; Baserga, S.J. Ribosomopathies: Old Concepts, New Controversies. Trends Genet. 2019, 35, 754–767. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.S.; Arnold, H.; Sun, X.X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26, 3332–3345, correction in 2009, 28, 993. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.M.; Zhou, X.; Gatignol, A.; Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 2014, 33, 4916–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgado-Palacin, L.; Varetti, G.; Llanos, S.; Gómez-López, G.; Martinez, D.; Serrano, M. Partial Loss of Rpl11 in Adult Mice Recapitulates Diamond-Blackfan Anemia and Promotes Lymphomagenesis. Cell Rep. 2015, 13, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, J. Ribosome heterogeneity in stem cells and development. J. Cell Biol. 2020, 219, e202001108. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.; Hopes, T.; Aspden, J.L. Ribosome heterogeneity and specialization in development. Wiley Interdiscip. Rev. RNA 2021, 12, e1644. [Google Scholar] [CrossRef]
- Favaro, F.P.; Alvizi, L.; Zechi-Ceide, R.M.; Bertola, D.; Felix, T.M.; de Souza, J.; Raskin, S.; Twigg, S.R.; Weiner, A.M.; Armas, P.; et al. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Hum. Genet. 2014, 94, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012, 44, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, J.A.; Traylor, R.N.; Schaefer, G.B.; McPherson, E.W.; Ballif, B.C.; Klopocki, E.; Mundlos, S.; Shaffer, L.G.; Aylsworth, A.S.; 1q21.1 Study Group. Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur. J. Hum. Genet. 2012, 20, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Pilaz, L.J.; McMahon, J.J.; Golzio, C.; Wu, D.; Shi, L.; Katsanis, N.; Silver, D.L. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 2015, 35, 7003–7018. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; McMahon, J.J.; Tsai, Y.H.; Wang, Z.; Silver, D.L. Haploinsufficiency for Core Exon Junction Complex Components Disrupts Embryonic Neurogenesis and Causes p53-Mediated Microcephaly. PLoS Genet. 2016, 12, e1006282. [Google Scholar] [CrossRef] [Green Version]
- Su, C.H.; Liao, W.J.; Ke, W.C.; Yang, R.B.; Tarn, W.Y. The Y14-p53 regulatory circuit in megakaryocyte differentiation and thrombocytopenia. bioRxiv 2020. [Google Scholar] [CrossRef]
- Silver, D.L.; Watkins-Chow, D.E.; Schreck, K.C.; Pierfelice, T.J.; Larson, D.M.; Burnetti, A.J.; Liaw, H.J.; Myung, K.; Walsh, C.A.; Gaiano, N.; et al. The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nat. Neurosci. 2010, 13, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Chuang, T.W.; Lu, C.C.; Su, C.H.; Wu, P.Y.; Easwvaran, S.; Lee, C.C.; Kuo, H.C.; Hung, K.Y.; Lee, K.M.; Tsai, C.Y.; et al. The RNA Processing Factor Y14 Participates in DNA Damage Response and Repair. iScience 2019, 13, 402–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.S.; Mishra, R.; Safieddine, A.; Coleno, E.; Alasseur, Q.; Faucourt, M.; Barbosa, I.; Bertrand, E.; Spassky, N.; Le Hir, H. Exon junction complex dependent mRNA localization is linked to centrosome organization during ciliogenesis. Nat. Commun. 2021, 12, 1351. [Google Scholar] [CrossRef]
- Lu, C.C.; Lee, C.C.; Tseng, C.T.; Tarn, W.Y. Y14 governs p53 expression and modulates DNA damage sensitivity. Sci. Rep. 2017, 7, 45558. [Google Scholar]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Jacobs, A.C.; Van Houten, B.; Greenberg, M.M. Double-strand break formation during nucleotide excision repair of a DNA interstrand cross-link. Biochemistry 2009, 48, 7565–7567. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [Green Version]
- García-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.T.; Berto, G.E.; Di Cunto, F. Impact of DNA repair and stability defects on cortical development. Cell Mol. Life Sci. 2018, 75, 3963–3976. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012, 11, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sii-Felice, K.; Etienne, O.; Hoffschir, F.; Mathieu, C.; Riou, L.; Barroca, V.; Haton, C.; Arwert, F.; Fouchet, P.; Boussin, F.D.; et al. Fanconi DNA repair pathway is required for survival and long-term maintenance of neural progenitors. EMBO J. 2008, 27, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Jacks, T. Double indemnity: p53, BRCA and cancer. p53 mutation partially rescues developmental arrest in Brca1 and Brca2 null mice, suggesting a role for familial breast cancer genes in DNA damage repair. Nat. Med. 1997, 3, 721–722. [Google Scholar] [CrossRef]
- Adam, J.; Deans, B.; Thacker, J. A role for Xrcc2 in the early stages of mouse development. DNA Repair 2007, 6, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Hasty, P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 1996, 16, 7133–7143. [Google Scholar] [CrossRef] [Green Version]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 2000, 5, 993–1002. [Google Scholar] [CrossRef]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.O.; Tong, W.M.; Demuth, I.; Radovanovic, I.; Herceg, Z.; Aguzzi, A.; Digweed, M.; Wang, Z.Q. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 2005, 11, 538–544. [Google Scholar] [CrossRef]
- Jaber, S.; Toufektchan, E.; Lejour, V.; Bardot, B.; Toledo, F. p53 downregulates the Fanconi anaemia DNA repair pathway. Nat. Commun. 2016, 7, 11091. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, V.; Lingner, J. Replication of telomeres and the regulation of telomerase. Cold Spring Harb. Perspect. Biol. 2013, 5, a010405. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186, correction in 2019, 20, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, Y.; He, J.; Kulkarni, S.; Bessler, M.; Mason, P.J. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. USA 2004, 101, 10756–10761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Rudnick, D.A.; He, J.; Crimmins, D.L.; Ladenson, J.H.; Bessler, M.; Mason, P.J. Dyskerin ablation in mouse liver inhibits rRNA processing and cell division. Mol. Cell Biol. 2010, 30, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereboeva, L.; Hubbard, M.; Goldman, F.D.; Westin, E.R. Robust DNA Damage Response and Elevated Reactive Oxygen Species in TINF2-Mutated Dyskeratosis Congenita Cells. PLoS ONE 2016, 11, e0148793. [Google Scholar] [CrossRef] [Green Version]
- Ferrón, S.R.; Marqués-Torrejón, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Fariñas, I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 2009, 29, 14394–14407. [Google Scholar] [CrossRef] [PubMed]
- Tejera, A.M.; Stagno d’Alcontres, M.; Thanasoula, M.; Marion, R.M.; Martinez, P.; Liao, C.; Flores, J.M.; Tarsounas, M.; Blasco, M.A. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev. Cell 2010, 18, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyelles, M.; Episkopou, H.; O’Donohue, M.F.; Kermasson, L.; Frange, P.; Poulain, F.; Burcu Belen, F.; Polat, M.; Bole-Feysot, C.; Langa-Vives, F.; et al. Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models. EMBO Mol. Med. 2019, 11, e10201. [Google Scholar] [CrossRef] [PubMed]
- Toufektchan, E.; Lejour, V.; Durand, R.; Giri, N.; Draskovic, I.; Bardot, B.; Laplante, P.; Jaber, S.; Alter, B.P.; Londono-Vallejo, J.A.; et al. Germline mutation of MDM4, a major p53 regulator, in a familial syndrome of defective telomere maintenance. Sci. Adv. 2020, 6, eaay3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeonova, I.; Jaber, S.; Draskovic, I.; Bardot, B.; Fang, M.; Bouarich-Bourimi, R.; Lejour, V.; Charbonnier, L.; Soudais, C.; Bourdon, J.C.; et al. Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep. 2013, 3, 2046–2058. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.; Collopy, L.; Cardoso, S.; de la Fuente, J.; Lawson, S.; Powell, J.; Cooper, N.; Foster, A.; Mohammed, S.; et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Investig. 2015, 125, 2151–2160. [Google Scholar] [CrossRef]
- Gönczy, P. Towards a molecular architecture of centriole assembly. Nat. Rev. Mol. Cell Biol. 2012, 13, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.R.; Krämer, A. Centrosome amplification, chromosomal instability and cancer: Mechanistic, clinical and therapeutic issues. Chromosome Res. 2016, 24, 105–126. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal Transduct. Target. Ther. 2020, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- LoMastro, G.M.; Holland, A.J. The Emerging Link between Centrosome Aberrations and Metastasis. Dev. Cell 2019, 49, 325–331. [Google Scholar] [CrossRef]
- Haren, L.; Remy, M.H.; Bazin, I.; Callebaut, I.; Wright, M.; Merdes, A. NEDD1-dependent recruitment of the gamma-tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J. Cell Biol. 2006, 172, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Goundiam, O.; Basto, R. Centrosomes in disease: How the same music can sound so different? Curr. Opin. Struct. Biol. 2021, 66, 74–82. [Google Scholar] [CrossRef]
- Siskos, N.; Stylianopoulou, E.; Skavdis, G.; Grigoriou, M.E. Molecular Genetics of Microcephaly Primary Hereditary: An Overview. Brain Sci. 2021, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.L.; Kosodo, Y.; Enard, W.; Pääbo, S.; Huttner, W.B. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10438–10443. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.E.; Garcia, I.; Crowther, A.J.; Li, S.; Stewart, A.; Liu, H.; Lough, K.J.; O’Neill, S.; Veleta, K.; Oyarzabal, E.A.; et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Development 2015, 142, 3921–3932. [Google Scholar] [CrossRef] [Green Version]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, F.T.; Tocco, C.; Pallavicini, G.; Liu, Y.; Vernì, F.; Merigliano, C.; Bonaccorsi, S.; El-Assawy, N.; Priano, L.; Gai, M.; et al. Citron Kinase Deficiency Leads to Chromosomal Instability and TP53-Sensitive Microcephaly. Cell Rep. 2017, 18, 1674–1686. [Google Scholar] [CrossRef]
- Klingseisen, A.; Jackson, A.P. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011, 25, 2011–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.J.; Marjanović, M.; Lüders, J.; Stracker, T.H.; Costanzo, V. Cep63 and cep152 cooperate to ensure centriole duplication. PLoS ONE 2013, 8, e69986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjanović, M.; Sánchez-Huertas, C.; Terré, B.; Gómez, R.; Scheel, J.F.; Pacheco, S.; Knobel, P.A.; Martínez-Marchal, A.; Aivio, S.; Palenzuela, L.; et al. CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat. Commun. 2015, 6, 7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monda, J.K.; Cheeseman, I.M. Nde1 promotes diverse dynein functions through differential interactions and exhibits an isoform-specific proteasome association. Mol. Biol. Cell 2018, 29, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.P.; Maryniak, A.L.; Boatwright, C.A.; Lee, J.; Atkins, A.; Tijhuis, A.; Spierings, D.C.; Bazzi, H.; Foijer, F.; Jordan, P.W.; et al. Centrosome defects cause microcephaly by activating the 53BP1-USP28-TP53 mitotic surveillance pathway. EMBO J. 2021, 40, e106118. [Google Scholar] [CrossRef]
- Ruggero, D.; Grisendi, S.; Piazza, F.; Rego, E.; Mari, F.; Rao, P.H.; Cordon-Cardo, C.; Pandolfi, P.P. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 2003, 299, 259–262. [Google Scholar] [CrossRef]
- Yoon, A.; Peng, G.; Brandenburger, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L. Turning Uridines around: Role of rRNA Pseudouridylation in Ribosome Biogenesis and Ribosomal Function. Biomolecules 2018, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, M.; Lindbæk, L.; Krogh, N.; Doganli, C.; Keller, C.; Mönnich, M.; Gonçalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A links primary microcephaly to dysfunction of ribosome biogenesis, resorption of primary cilia, and neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef]
- Smith, E.; Dejsuphong, D.; Balestrini, A.; Hampel, M.; Lenz, C.; Takeda, S.; Vindigni, A.; Costanzo, V. An ATM- and ATR-dependent checkpoint inactivates spindle assembly by targeting CEP63. Nat. Cell Biol. 2009, 11, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Nalepa, G.; Enzor, R.; Sun, Z.; Marchal, C.; Park, S.J.; Yang, Y.; Tedeschi, L.; Kelich, S.; Hanenberg, H.; Clapp, D.W. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J. Clin. Investig. 2013, 123, 3839–3847. [Google Scholar] [CrossRef] [Green Version]
- Gueiderikh, A.; Maczkowiak-Chartois, F.; Rouvet, G.; Souquère-Besse, S.; Apcher, S.; Diaz, J.J.; Rosselli, F. Fanconi anemia A protein participates in nucleolar homeostasis maintenance and ribosome biogenesis. Sci. Adv. 2021, 7, eabb5414. [Google Scholar] [CrossRef]
- Sohn, D.; Graupner, V.; Neise, D.; Essmann, F.; Schulze-Osthoff, K.; Jänicke, R.U. Pifithrin-alpha protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell Death Differ. 2009, 16, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Macari, E.R.; Chan, I.T.; Blair, M.C.; Doulatov, S.; Vo, L.T.; Raiser, D.M.; Siva, K.; Basak, A.; Pirouz, M.; et al. Calmodulin inhibitors improve erythropoiesis in Diamond-Blackfan anemia. Sci. Transl. Med. 2020, 12, eabb5831. [Google Scholar] [CrossRef] [PubMed]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer therapeutic targeting using mutant-p53-specific siRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [Green Version]
- Jaako, P.; Debnath, S.; Olsson, K.; Bryder, D.; Flygare, J.; Karlsson, S. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 2012, 120, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Payne, E.M.; Virgilio, M.; Narla, A.; Sun, H.; Levine, M.; Paw, B.H.; Berliner, N.; Look, A.T.; Ebert, B.L.; Khanna-Gupta, A. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012, 120, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Dixon, J.; Achilleos, A.; Dixon, M.; Trainor, P.A. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat. Commun. 2016, 7, 10328. [Google Scholar] [CrossRef] [Green Version]
- de Peralta, M.S.; Mouguelar, V.S.; Sdrigotti, M.A.; Ishiy, F.A.; Fanganiello, R.D.; Passos-Bueno, M.R.; Coux, G.; Calcaterra, N.B. Cnbp ameliorates Treacher Collins Syndrome craniofacial anomalies through a pathway that involves redox-responsive genes. Cell Death Dis. 2016, 7, e2397. [Google Scholar] [CrossRef] [Green Version]
- Rosas, M.G.; Lorenzatti, A.; Porcel de Peralta, M.S.; Calcaterra, N.B.; Coux, G. Proteasomal inhibition attenuates craniofacial malformations in a zebrafish model of Treacher Collins Syndrome. Biochem. Pharmacol. 2019, 163, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 375, 1095. [Google Scholar] [PubMed]
- Zhou, L.; McMahon, C.; Bhagat, T.; Alencar, C.; Yu, Y.; Fazzari, M.; Sohal, D.; Heuck, C.; Gundabolu, K.; Ng, C.; et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011, 71, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-β Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Río, P.; Navarro, S.; Bueren, J.A. Advances in Gene Therapy for Fanconi Anemia. Hum. Gene Ther. 2018, 29, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Sanz, E.; Bean, J.C.; Carey, D.P.; Quintana, A.; McKnight, G.S. RiboTag: Ribosomal Tagging Strategy to Analyze Cell-Type-Specific mRNA Expression In Vivo. Curr. Protoc. Neurosci. 2019, 88, e77. [Google Scholar] [CrossRef]
- Ribezzo, F.; Snoeren, I.A.M.; Ziegler, S.; Stoelben, J.; Olofsen, P.A.; Henic, A.; Ferreira, M.V.; Chen, S.; Stalmann, U.S.A.; Buesche, G.; et al. Rps14, Csnk1a1 and miRNA145/miRNA146a deficiency cooperate in the clinical phenotype and activation of the innate immune system in the 5q- syndrome. Leukemia 2019, 33, 1759–1772. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-Y.; Su, C.-H.; Tarn, W.-Y. p53 Activation in Genetic Disorders: Different Routes to the Same Destination. Int. J. Mol. Sci. 2021, 22, 9307. https://doi.org/10.3390/ijms22179307
Tsai Y-Y, Su C-H, Tarn W-Y. p53 Activation in Genetic Disorders: Different Routes to the Same Destination. International Journal of Molecular Sciences. 2021; 22(17):9307. https://doi.org/10.3390/ijms22179307
Chicago/Turabian StyleTsai, Yu-Young, Chun-Hao Su, and Woan-Yuh Tarn. 2021. "p53 Activation in Genetic Disorders: Different Routes to the Same Destination" International Journal of Molecular Sciences 22, no. 17: 9307. https://doi.org/10.3390/ijms22179307
APA StyleTsai, Y. -Y., Su, C. -H., & Tarn, W. -Y. (2021). p53 Activation in Genetic Disorders: Different Routes to the Same Destination. International Journal of Molecular Sciences, 22(17), 9307. https://doi.org/10.3390/ijms22179307