
Pharmaco-Magnetic Resonance as a Tool for Monitoring the Medication-Related Effects in the Brain May Provide Potential Biomarkers for Psychotic Disorders



Abstract
:1. Introduction
2. Methods
3. Results
3.1. Etiological Theories of Schizophrenia
3.1.1. Neurodevelopment vs. Neurodegeneration
3.1.2. Biochemical Explanation of Psychosis
3.1.3. Schizophrenia as a Syndrome of Impaired Functional Brain Connectivity
3.1.4. An Integral Etiological Framework for Schizophrenia
3.2. The Role of Neuroimaging and Translational Neuroscience in Schizophrenia Research
3.3. Implications of Neuroimaging Findings for the Treatment of Schizophrenia
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gaebel, W.; Zielasek, J. Focus on psychosis. Dialog. Clin. Neurosci. 2015, 17, 9–18. [Google Scholar]
- Rössler, W.; Salize, H.J.; van Os, J.; Riecher-Rössler, A. Size of burden of schizophrenia and psychotic disorders. Eur. Neuropsychopharmacol. 2005, 15, 399–409. [Google Scholar] [CrossRef]
- Joyce, E.M.; Roiser, J. Cognitive heterogeneity in schizophrenia. Curr. Opin. Psychiatry 2007, 20, 268–272. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Addington, J.; Heinssen, R. Prediction and Prevention of Psychosis in Youth at Clinical High Risk. Annu. Rev. Clin. Psychol. 2012, 8, 269–289. [Google Scholar] [CrossRef] [Green Version]
- Freudenreich, O. Differential Diagnosis of Psychotic Symptoms: Medical “Mimics”. Psychiatr. Times 2010, 27, 56–61. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Association Publishing: Washington, DC, USA, 2013. [Google Scholar]
- World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines; World Health Organization: Geneva, Switzerland, 1992. [Google Scholar]
- Jakobsen, K.D.; Frederiksen, J.N.; Hansen, T.F.; Jansson, L.B.; Parnas, J.; Werge, T. Reliability of clinical ICD-10 schizophrenia diagnoses. Nord. J. Psychiatry 2005, 59, 209–212. [Google Scholar] [CrossRef]
- Brunoni, A.R. Beyond the DSM: Trends in psychiatry diagnoses. Rev. Psiquiatr. Clin. 2017, 44, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Trossbach, S.V.; Hecher, L.; Schafflick, D.; Deenen, R.; Popa, O.; Lautwein, T.; Tschirner, S.; Köhrer, K.; Fehsel, K.; Papazova, I.; et al. Dysregulation of a specific immune-related network of genes biologically defines a subset of schizophrenia. Transl. Psychiatry 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanov, D.S. An Essay on the Mind-Brain Problem and Legal Proof. Balk. J. Philos. 2018, 10, 27–36. [Google Scholar] [CrossRef]
- Brewer, L. General Psychology: Required Reading; Deiner Education Fund: Salt Lake City, CT, USA, 2019; p. 323. [Google Scholar]
- Bender, L. Childhood schizophrenia: Clinical study of one hundred schizophrenic children. Am. J. Orthopsychiatry 1947, 17, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.J.; Mortensen, E.L.; Schiffman, J.; Reinisch, J.M.; Maeda, J.; Mednick, S.A. Early developmental milestones and risk of schizophrenia: A 45-year follow-up of the Copenhagen Perinatal Cohort. Schizophr. Res. 2010, 118, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, I.M.; Bearden, C.; Hollister, J.M.; Gasperoni, T.L.; Sanchez, L.E.; Hadley, T.; Cannon, T.D. Childhood Neuromotor Dysfunction in Schizophrenia Patients and Their Unaffected Siblings: A Prospective Cohort Study. Schizophr. Bull. 2000, 26, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Jones, P. Child developmental risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet 1994, 344, 1398–1402. [Google Scholar] [CrossRef]
- Ollendick, T.H.; Prinz, R.J. (Eds.) Advances in Clinical Child Psychology; Springer US: New York, NY, USA, 1996; Volume 18. [Google Scholar]
- Ridler, K.; Veijola, J.M.; Tanskanen, P.; Miettunen, J.; Chitnis, X.; Suckling, J.; Murray, G.; Haapea, M.; Jones, P.B.; Isohanni, M.K.; et al. Fronto-cerebellar systems are associated with infant motor and adult executive functions in healthy adults but not in schizophrenia. Proc. Natl. Acad. Sci. USA 2006, 103, 15651–15656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taanila, A.; Murray, G.; Jokelainen, J.; Isohanni, M.; Rantakallio, P. Infant developmental milestones: A 31-year follow-up. Dev. Med. Child Neurol. 2005, 47, 581. [Google Scholar] [CrossRef]
- Kremen, W.S.; Vinogradov, S.; Poole, J.H.; Schaefer, C.A.; Deicken, R.F.; Factor-Litvak, P.; Brown, A.S. Cognitive decline in schizophrenia from childhood to midlife: A 33-year longitudinal birth cohort study. Schizophr. Res. 2010, 118, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.; Moffitt, T.E.; Caspi, A.; Murray, R.M.; Harrington, H.; Poulton, R. Neuropsychological performance at the age of 13 years and adult schizophreniform disorder: Prospective birth cohort study. Br. J. Psychiatry 2006, 189, 463–464. [Google Scholar] [CrossRef] [Green Version]
- Caspi, A.; Reichenberg, A.; Weiser, M.; Rabinowitz, J.; Kaplan, Z.; Knobler, H.; Davidson-Sagi, N.; Davidson, M. Cognitive performance in schizophrenia patients assessed before and following the first psychotic episode. Schizophr. Res. 2003, 65, 87–94. [Google Scholar] [CrossRef]
- Kobayashi, H.; Isohanni, M.; Jääskeläinen, E.; Miettunen, J.; Veijola, J.; Haapea, M.; Järvelin, M.-R.; Jones, P.B.; Murray, G.K. Linking the developmental and degenerative theories of schizophrenia: Association between infant development and adult cognitive decline. Schizophr. Bull. 2014, 40, 1319–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulshoff Pol, H.E.; Kahn, R.S. What Happens After the First Episode? A Review of Progressive Brain Changes in Chronically Ill Patients with Schizophrenia. Schizophr. Bull. 2008, 34, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Olabi, B.; Ellison-Wright, I.; McIntosh, A.; Wood, S.; Bullmore, E.; Lawrie, S. Are There Progressive Brain Changes in Schizophrenia? A Meta-Analysis of Structural Magnetic Resonance Imaging Studies. Biol. Psychiatry 2011, 70, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Kempton, M.J.; Stahl, D.; Williams, S.; DeLisi, L.E. Progressive lateral ventricular enlargement in schizophrenia: A meta-analysis of longitudinal MRI studies. Schizophr. Res. 2010, 120, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, J.A. Is schizophrenia a neurodegenerative disorder? a clinical and neurobiological perspective. Biol. Psychiatry 1999, 46, 729–739. [Google Scholar] [CrossRef]
- Lin, Y.; Li, M.; Zhou, Y.; Deng, W.; Ma, X.; Wang, Q.; Guo, W.; Li, Y.; Jiang, L.; Hu, X.; et al. Age-Related Reduction in Cortical Thickness in First-Episode Treatment-Naïve Patients with Schizophrenia. Neurosci. Bull. 2019, 35, 688–696. [Google Scholar] [CrossRef]
- Walker, E.F.; Lewine, R.R.; Neumann, C. Childhood behavioral characteristics and adult brain morphology in schizophrenia. Schizophr. Res. 1996, 22, 93–101. [Google Scholar] [CrossRef]
- Seeman, P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987, 1, 133–152. [Google Scholar] [CrossRef]
- Harrington, A. The fall of the schizophrenogenic mother. Lancet 2012, 379, 1292–1293. [Google Scholar] [CrossRef]
- Johnston, J. The Ghost of the Schizophrenogenic Mother. Virtual Mentor 2013, 15, 801–805. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Stahl, S.M. The Dopamine Hypothesis of Schizophrenia: A Review. Schizophr. Bull. 1976, 2, 19–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, A.; Lindqvist, M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol. Toxicol. 1963, 20, 140–144. [Google Scholar] [CrossRef]
- Seeman, P.; Lee, T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975, 188, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Kane, J.M.; Alvir, J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology 1987, 91, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Su, T.-P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef]
- Abi-Dargham, A.; Gil, R.; Krystal, J.; Baldwin, R.M.; Seibyl, J.P.; Bowers, M.; Van Dyck, C.H.; Charney, D.S.; Innis, R.B.; Laruelle, M. Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am. J. Psychiatry 1998, 155, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Kegeles, L.S.; Abi-Dargham, A.; Zea-Ponce, Y.; Rodenhiser-Hill, J.; Mann, J.; Van Heertum, R.L.; Cooper, T.B.; Carlsson, A.; Laruelle, M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biol. Psychiatry 2000, 48, 627–640. [Google Scholar] [CrossRef]
- Gibbs, F.A. Ictal and non-ictal psychiatric disorders in temporal lobe epilepsy. J. Nerv. Ment. Dis. 1951, 113, 522–528. [Google Scholar]
- Malamud, N. Psychiatric Disorder With Intracranial Tumors of Limbic System. Arch. Neurol. 1967, 17, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Javitt, D.C. Tinking Glutamatergically: Changing Concepts of Schizophrenia Based Upon Changing Neurochemical Models. Clin. Schizophr. Relat. Psychoses 2010, 4, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.C.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. [Google Scholar] [CrossRef]
- Lahti, A.C.; Koffel, B.; LaPorte, D.; Tamminga, C.A. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 1995, 13, 9–19. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D2 and serotonin 5-HT2 receptors—Implications for models of schizophrenia. Mol. Psychiatry 2002, 7, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohlich, J.; Van Horn, J.D. Reviewing the ketamine model for schizophrenia. J. Psychopharmacol. 2014, 28, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Glutamate and Schizophrenia: Phencyclidine, N-Methyl-d-Aspartate Receptors, and Dopamine–Glutamate Interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Farber, N.B. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007. [Google Scholar] [CrossRef]
- Grace, A. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 1991, 41, 1–24. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S.; Selemon, L.D. Functional and Anatomical Aspects of Prefrontal Pathology in Schizophrenia. Schizophr. Bull. 1997, 23, 437–458. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, D.R.; Berman, K.F. Prefrontal function in schizophrenia: Confounds and controversies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1996, 351, 1495–1503. [Google Scholar] [CrossRef]
- Du, X.; Choa, F.-S.; Chiappelli, J.; Wisner, K.M.; Wittenberg, G.; Adhikari, B.; Bruce, H.; Rowland, L.M.; Kochunov, P.; Hong, L.E. Aberrant Middle Prefrontal-Motor Cortex Connectivity Mediates Motor Inhibitory Biomarker in Schizophrenia. Biol. Psychiatry 2019, 85, 49–59. [Google Scholar] [CrossRef]
- Walton, E.; Hibar, D.P.; Van Erp, T.G.M.; Potkin, S.G.; Roiz-Santiañez, R.; Crespo-Facorro, B.; Suarez-Pinilla, P.; Van Haren, N.E.M.; De Zwarte, S.M.C.; Kahn, R.S.; et al. Prefrontal cortical thinning links to negative symptoms in schizophrenia via the ENIGMA consortium. Psychol. Med. 2018, 48, 82–94. [Google Scholar] [CrossRef]
- Robinson, T.E.; Becker, J.B. Enduring changes in brain and behavior produced by chronic amphetamine administration: A review and evaluation of animal models of amphetamine psychosis. Brain Res. Rev. 1986, 11, 157–198. [Google Scholar] [CrossRef]
- Schmidt, W.J.; Beninger, R.J. Behavioural Sensitization in Addiction, Schizophrenia, Parkinson’s Disease and Dyskinesia. Neurotox. Res. 2006, 10, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Wearne, T.A.; Cornish, J.L. Inhibitory regulation of the prefrontal cortex following behavioral sensitization to amphetamine and/or methamphetamine psychostimulants: A review of GABAergic mechanisms. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 95, 109681. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kornhuber, H.; Schmid-Burgk, W.; Holzmüller, B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Wachtel, H.; Turski, L. Glutamate: A new target in schizophrenia? Trends Pharmacol. Sci. 1990, 11, 219–220. [Google Scholar] [CrossRef]
- Coyle, J.T. NMDA Receptor and Schizophrenia: A Brief History. Schizophr. Bull. 2012, 38, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Mastropaolo, J.; Schwartz, B.L.; Rosse, R.B.; Morihisa, J.M. A “glutamatergic hypothesis” of schizophrenia. Rationale for pharmacotherapy with glycine. Clin. Neuropharmacol. 1989, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kornhuber, H.; Brand, U.; Menge, H. Effects of chronic amphetamine treatment on the glutamate concentration in cerebrospinal fluid and brain: Implications for a theory of schizophrenia. Neurosci. Lett. 1981, 24, 93–96. [Google Scholar] [CrossRef]
- Swerdlow, N.R. (Ed.) Integrative Circuit Models and Their Implications for the Pathophysiologies and Treatments of the Schizophrenias. In Behavioral Neurobiology of Schizophrenia and Its Treatment; Springer: Berlin/Heidelberg, Germany, 2010; Volume 4, pp. 555–583. [Google Scholar]
- Coyle, J.T. The Glutamatergic Dysfunction Hypothesis for Schizophrenia. Harv. Rev. Psychiatry 1996, 3, 241–253. [Google Scholar] [CrossRef]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, D.R. A connectionist approach to the prefrontal cortex. J. Neuropsychiatry Clin. Neurosci. 1993, 5, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Friston, K.; Frith, C.D. Schizophrenia: A disconnection syndrome? Clin. Neurosci. 1995, 3, 89–97. [Google Scholar]
- Friston, K.; Brown, H.R.; Siemerkus, J.; Stephan, K.E. The dysconnection hypothesis (2016). Schizophr. Res. 2016, 176, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Friston, K. Theoretical neurobiology and schizophrenia. Br. Med. Bull. 1996, 52, 644–655. [Google Scholar] [CrossRef]
- Bolton, T.A.W.; Wotruba, D.; Buechler, R.; Theodoridou, A.; Michels, L.; Kollias, S.; Rössler, W.; Heekeren, K.; Van De Ville, D. Triple Network Model Dynamically Revisited: Lower Salience Network State Switching in Pre-psychosis. Front. Physiol. 2020, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Menon, V. Salience Network. Brain Mapping; Elsevier: Amsterdam, The Netherlands, 2015; pp. 597–611. [Google Scholar]
- Buckner, R.L. The serendipitous discovery of the brain’s default network. NeuroImage 2012, 62, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, G.; Zeighami, Y.; Clark, C.A.; Coull, J.T.; Nagano-Saito, A.; Leyton, M.; Dagher, A.; Mišić, B. Dopamine Signaling Modulates the Stability and Integration of Intrinsic Brain Networks. Cereb. Cortex 2019, 29, 397–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckner, R.L.; Andrews-Hanna, E.J.R.; Schactera, D.L. The brain’s default network: Anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 2008. [Google Scholar]
- Greicius, M.D.; Krasnow, B.; Reiss, A.L.; Menon, V. Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Uddin, L.Q.; Yeo, B.T.T.; Spreng, R.N. Towards a Universal Taxonomy of Macro-scale Functional Human Brain Networks. Brain Topogr. 2019, 32, 926–942. [Google Scholar] [CrossRef]
- Krmpotich, T.D.; Tregellas, J.R.; Thompson, L.L.; Banich, M.T.; Klenk, A.M.; Tanabe, J.L. Resting-state activity in the left executive control network is associated with behavioral approach and is increased in substance dependence. Drug Alcohol Depend. 2013, 129, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, W.W.; Menon, V.; Schatzberg, A.F.; Keller, J.; Glover, G.H.; Kenna, H.; Reiss, A.L.; Greicius, M.D. Dissociable Intrinsic Connectivity Networks for Salience Processing and Executive Control. J. Neurosci. 2007, 27, 2349–2356. [Google Scholar] [CrossRef]
- Fox, M.D.; Snyder, A.Z.; Vincent, J.L.; Corbetta, M.; Van Essen, D.C.; Raichle, M.E. From The Cover: The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. USA 2005, 102, 9673–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, J.R.; Frost, J.A.; Hammeke, T.A.; Bellgowan, P.S.; Rao, S.M.; Cox, R.W. Conceptual processing during the conscious resting state. A functional MRI study. J. Cogn. Neurosci. 1999, 11, 80–95. [Google Scholar] [CrossRef]
- Menon, V. Large-scale brain networks and psychopathology: A unifying triple network model. Trends Cogn. Sci. 2011, 15, 483–506. [Google Scholar] [CrossRef]
- Williamson, P. Are Anticorrelated Networks in the Brain Relevant to Schizophrenia? Schizophr. Bull. 2007, 33, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Glutamate and dopamine dysregulation in schizophrenia—A synthesis and selective review. J. Psychopharmacol. 2007, 21, 440–452. [Google Scholar] [CrossRef]
- Olney, J.W.; Newcomer, J.W.; Farber, N. NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 1999, 33, 523–533. [Google Scholar] [CrossRef]
- Bleuler, E. Dementia Praecox or the Group of Schizophrenias; International Universities Press: Oxford, UK, 1950; p. 548. [Google Scholar]
- Weinberger, D.R. Implications of Normal Brain Development for the Pathogenesis of Schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669. [Google Scholar] [CrossRef]
- Murray, R.; Lewis, S. Is schizophrenia a neurodevelopmental disorder? Br. Med. J. Clin. Res. Ed. 1987, 295, 681–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulhara, P.; Gupta, S. What is schizophrenia: A neurodevelopmental or neurodegenerative disorder or a combination of both? A critical analysis. Indian J. Psychiatry 2010, 52, 21–27. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S.; Leranth, C.; Williams, S.M.; Mons, N.; Geffard, M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc. Natl. Acad. Sci. USA 1989, 86, 9015–9019. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Carvalho, F.; Eliez, S.; Caroni, P. Long-Lasting Rescue of Network and Cognitive Dysfunction in a Genetic Schizophrenia Model. Cell 2019, 178, 1387–1402. [Google Scholar] [CrossRef]
- Perkins, D.O.; Gu, H.; Boteva, K.; Lieberman, J.A. Relationship Between Duration of Untreated Psychosis and Outcome in First-Episode Schizophrenia: A Critical Review and Meta-Analysis. Am. J. Psychiatry 2005, 162, 1785–1804. [Google Scholar] [CrossRef] [PubMed]
- Aryutova, K.; Paunova, R.; Kandilarova, S.; Todeva-Radneva, A.; Stoyanov, D. Implications from translational cross-validation of clinical assessment tools for diagnosis and treatment in psychiatry. World J. Psychiatry 2021, 11, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Zhu, J.; Wang, C.; Qu, H.; Ma, X.; Tian, H.; Liu, M.; Qin, W. Brain structural and functional dissociated patterns in schizophrenia. BMC Psychiatry 2017, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Van Erp, T.G.M.; Hibar, D.P.; Rasmussen, J.M.; Glahn, D.C.; Pearlson, G.D.; Andreassen, O.A.; Agartz, I.; Westlye, L.T.; Haukvik, U.K.; Dale, A.M.; et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol. Psychiatry 2016, 21, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Womer, F.Y.; Bai, C.; Zhou, Q.; Wei, S.; Jiang, X.; Geng, H.; Zhou, Y.; Tang, Y.; Wang, F. Voxel-Based Morphometry in Individuals at Genetic High Risk for Schizophrenia and Patients with Schizophrenia during Their First Episode of Psychosis. PLoS ONE 2016, 11, e0163749. [Google Scholar] [CrossRef]
- Wright, I.C.; Rabe-Hesketh, S.; Woodruff, P.W.; David, A.S.; Murray, R.; Bullmore, E. Meta-Analysis of Regional Brain Volumes in Schizophrenia. Am. J. Psychiatry 2000, 157, 16–25. [Google Scholar] [CrossRef]
- Stoyanov, D.; Kandilarova, S.; Aryutova, K.; Paunova, R.; Todeva-Radneva, A.; Latypova, A.; Kherif, F. Multivariate Analysis of Structural and Functional Neuroimaging Can Inform Psychiatric Differential Diagnosis. Diagnostics 2021, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.M.; Matheson, S.L.; Laurens, K.; Carr, V.J.; Green, M. Systematic Meta-Analysis of Insula Volume in Schizophrenia. Biol. Psychiatry 2012, 72, 775–784. [Google Scholar] [CrossRef]
- Goodkind, M.; Eickhoff, S.B.; Oathes, D.; Jiang, Y.; Chang, A.; Jones-Hagata, L.B.; Ortega, B.N.; Zaiko, Y.V.; Roach, E.L.; Korgaonkar, M.; et al. Identification of a Common Neurobiological Substrate for Mental Illness. JAMA Psychiatry 2015, 72, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, J.M.; Rogers, B.P.; Blackford, J.U.; Heckers, S.; Woodward, N.D. Insula functional connectivity in schizophrenia. Schizophr. Res. 2020, 220, 69–77. [Google Scholar] [CrossRef]
- Lee, S.-H.; Niznikiewicz, M.; Asami, T.; Otsuka, T.; Salisbury, D.F.; Shenton, M.E.; McCarley, R.W. Initial and Progressive Gray Matter Abnormalities in Insular Gyrus and Temporal Pole in First-Episode Schizophrenia Contrasted With First-Episode Affective Psychosis. Schizophr. Bull. 2016, 42, 790–801. [Google Scholar] [CrossRef]
- Mier, D.; Lis, S.; Zygrodnik, K.; Sauer, C.; Ulferts, J.; Gallhofer, B.; Kirsch, P. Evidence for altered amygdala activation in schizophrenia in an adaptive emotion recognition task. Psychiatry Res. 2014, 221, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goghari, V.M.; Sanford, N.; Spilka, M.J.; Woodward, T.S. Task-Related Functional Connectivity Analysis of Emotion Discrimination in a Family Study of Schizophrenia. Schizophr. Bull. 2017, 43, 1348–1362. [Google Scholar] [CrossRef]
- Belge, J.-B.; Maurage, P.; Mangelinckx, C.; LeLeux, D.; Delatte, B.; Constant, E. Facial decoding in schizophrenia is underpinned by basic visual processing impairments. Psychiatry Res. 2017, 255, 167–172. [Google Scholar] [CrossRef]
- Whitfield-Gabrieli, S.; Thermenos, H.W.; Milanovic, S.; Tsuang, M.T.; Faraone, S.; McCarley, R.; Shenton, M.E.; Green, A.I.; Nieto-Castanon, A.; LaViolette, P.; et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc. Natl. Acad. Sci. USA 2009, 106, 1279–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanov, D.; Aryutova, K.; Kandilarova, S.; Paunova, R.; Arabadzhiev, Z.; Todeva-Radneva, A.; Kostianev, S.; Borgwardt, S. Diagnostic Task Specific Activations in Functional MRI and Aberrant Connectivity of Insula with Middle Frontal Gyrus Can Inform the Differential Diagnosis of Psychosis. Diagnostics 2021, 11, 95. [Google Scholar] [CrossRef]
- Nekovarova, T.; Fajnerova, I.; Horáček, J.; Spaniel, F. Bridging disparate symptoms of schizophrenia: A triple network dysfunction theory. Front. Behav. Neurosci. 2014, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Pu, W.; Wang, J.; Liu, H.; Wu, G.; Liu, C.; Mwansisya, T.E.; Tao, H.; Chen, X.; Huang, X.; et al. Inefficient DMN Suppression in Schizophrenia Patients with Impaired Cognitive Function but not Patients with Preserved Cognitive Function. Sci. Rep. 2016, 6, 21657. [Google Scholar] [CrossRef] [Green Version]
- Aryutova, K.; Paunova, R.; Kandilarova, S.; Maes, M.; Stoyanova, K.; Stoyanov, D. Differential Aberrant Connectivity of Precuneus and Anterior Insula May Underpin the Diagnosis of Schizophrenia and Mood Disorders. WJP 2021, in press. [Google Scholar]
- Modinos, G.; Costafreda, S.G.; van Tol, M.-J.; McGuire, P.; Aleman, A.; Allen, P. Neuroanatomy of auditory verbal hallucinations in schizophrenia: A quantitative meta-analysis of voxel-based morphometry studies. Cortex 2013, 49, 1046–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, R.C.K.; Di, X.; McAlonan, G.; Gong, Q. Brain Anatomical Abnormalities in High-Risk Individuals, First-Episode, and Chronic Schizophrenia: An Activation Likelihood Estimation Meta-analysis of Illness Progression. Schizophr. Bull. 2011, 37, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Von Zerssen, D.; Koeller, D.M. Paranoid-Depressivitäts-Skala (PD-S); Beltz: Weinheim, Germany, 1976. [Google Scholar]
- Pearlson, G.D.; Calhoun, V. Structural and Functional Magnetic Resonance Imaging in Psychiatric Disorders. Can. J. Psychiatry 2007, 52, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, V.; Uddin, L.Q. Saliency, switching, attention and control: A network model of insula function. Brain Struct. Funct. 2010, 214, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Goulden, N.; Khusnulina, A.; Davis, N.J.; Bracewell, R.M.; Bokde, A.L.; McNulty, J.P.; Mullins, P.G. The salience network is responsible for switching between the default mode network and the central executive network: Replication from DCM. NeuroImage 2014, 99, 180–190. [Google Scholar] [CrossRef]
- Holtzheimer, P.E.; Mayberg, H.S. Deep Brain Stimulation for Psychiatric Disorders. Annu. Rev. Neurosci. 2011, 34, 289–307. [Google Scholar] [CrossRef] [Green Version]
- Khalili-Mahani, N.; Rombouts, S.A.; Van Osch, M.J.; Duff, E.P.; Carbonell, F.; Nickerson, L.D.; Becerra, L.; Dahan, A.; Evans, A.C.; Soucy, J.-P.; et al. Biomarkers, designs, and interpretations of resting-state fMRI in translational pharmacological research: A review of state-of-the-Art, challenges, and opportunities for studying brain chemistry. Hum. Brain Mapp. 2017, 38, 2276–2325. [Google Scholar] [CrossRef]
- Vollenweider, F.; Leenders, K.; Scharfetter, C.; Antonini, A.; Maguire, P.; Missimer, J.; Angst, J. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur. Neuropsychopharmacol. 1997, 7, 9–24. [Google Scholar] [CrossRef]
- Lahti, A.C.; Holcomb, H.H.; Medoff, D.R.; Tamminga, C.A. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. NeuroReport 1995, 6, 869–872. [Google Scholar] [CrossRef]
- Hartvig, P.; Valtysson, J.; Lindner, K.-J.; Kristensen, J.; Karlsten, R.; Gustafsson, L.L.; Persson, J.; Svensson, J.O.; Øye, I.; Antoni, G.; et al. Central nervous system effects of subdissociative doses of (S)-ketamine are related to plasma and brain concentrations measured with positron emission tomography in healthy volunteers. Clin. Pharmacol. Ther. 1995, 58, 165–173. [Google Scholar] [CrossRef]
- Fowler, J.; MacGregor, R.; Wolf, A.; Arnett, C.; Dewey, S.; Schlyer, D.; Christman, D.; Logan, J.; Smith, M.; Sachs, H.; et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 1987, 235, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Logan, J.; Gatley, S.J.; Dewey, S.L.; MacGregor, R.R.; Schlyer, D.J.; Pappas, N.; King, P.; Wang, G.J. Carbon-11-cocaine binding compared at subpharmacological and pharmacological doses: A PET study. J. Nucl. Med. 1995, 36, 1289–1297. [Google Scholar]
- Jenkins, B.G. Pharmacologic magnetic resonance imaging (phMRI): Imaging drug action in the brain. NeuroImage 2012, 62, 1072–1085. [Google Scholar] [CrossRef] [Green Version]
- Wandschneider, B.; Koepp, M.J. Pharmaco fMRI: Determining the functional anatomy of the effects of medication. NeuroImage Clin. 2016, 12, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Borsook, D.; Becerra, L.; Hargreaves, R. A role for fMRI in optimizing CNS drug development. Nat. Rev. Drug Discov. 2006, 5, 411–425. [Google Scholar] [CrossRef]
- Nathan, P.J.; Phan, K.L.; Harmer, C.J.; Mehta, M.A.; Bullmore, E.T. Increasing pharmacological knowledge about human neurological and psychiatric disorders through functional neuroimaging and its application in drug discovery. Curr. Opin. Pharmacol. 2014, 14, 54–61. [Google Scholar] [CrossRef]
- Stoianov, D.S. Validation Theory-from basic neuroscience to pharmacopsychology. S. Afr. J. Psychiatry 2007, 13, 116. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concept | Definition | Studies | Main Findings/Conclusion |
|---|---|---|---|
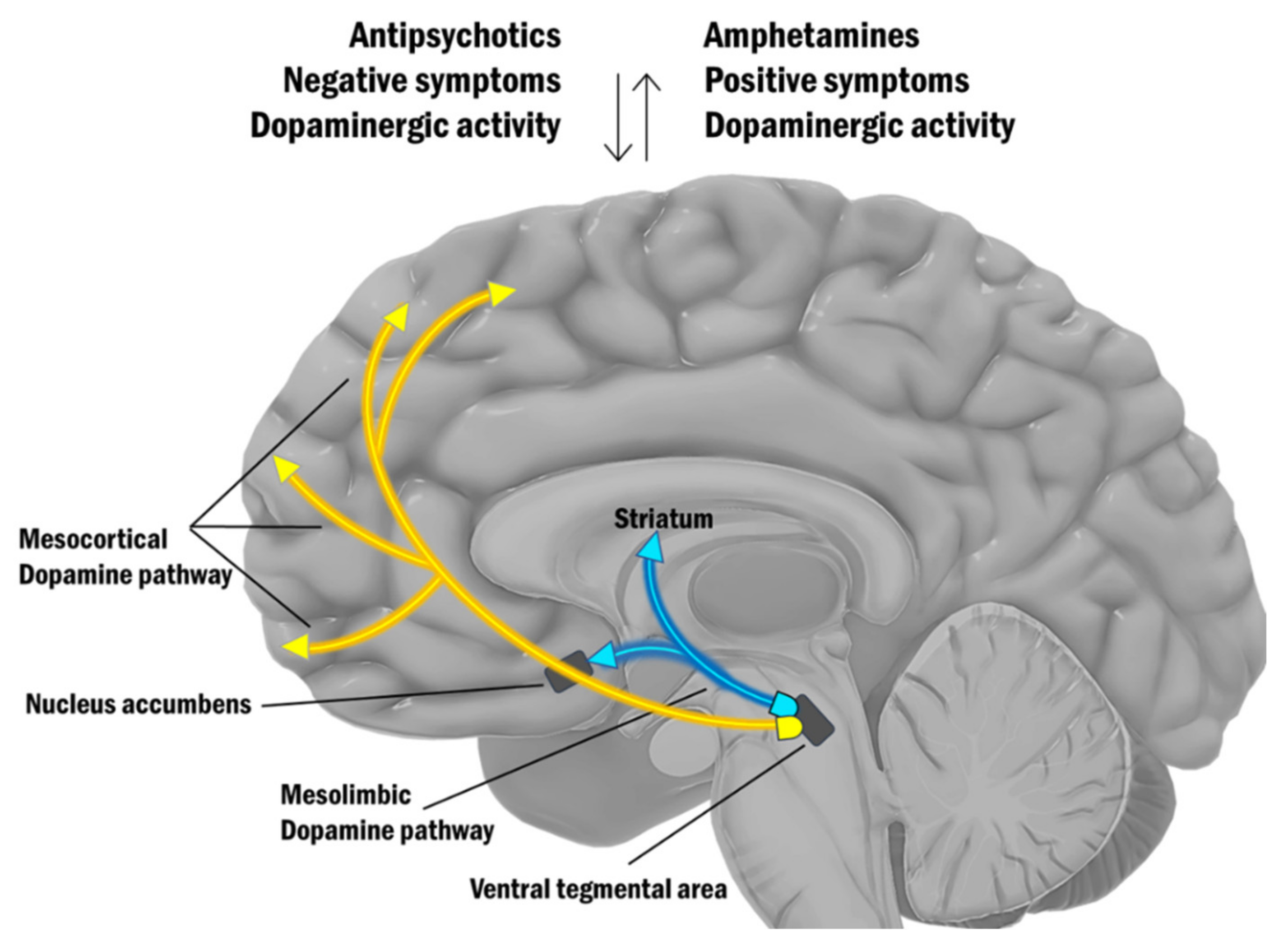
| Dopamine hypothesis | Hyperdopaminergia in the mesolimbic system causes psychotic symptoms and hypodopaminergia in the mesocortical pathway is the reason for negative symptoms. | Seeman and Lee (1975) [36] | Neuroleptics’ efficacy in blocking dopamine D2 receptors. |
| Lieberman et al. (1987) [37] | Psychotic phenomena are triggered by the dopamine agonists. | ||
| Breier et al. (1997) [38] Laruelle et al. (1999) [39] Abi-Dargham et al. (1998) [40] Kegeles et al. (2000) [41] | Amphetamine-induced hyperdopaminergia in the striatum is significantly higher in patients with schizophrenia compared to healthy controls. | ||
| Gibbs (1951) [42] | Epileptic seizures caused by lesions in the limbic regions lead to florid psychotic production. | ||
| Malamud (1967) [43] | Tumors in the limbic structures cause psychotic symptoms. | ||
| Glutamate hypothesis | A decrease of glutamate activity at the glutamate synapse, particularly in the prefrontal cortex induces positive, negative, and cognitive symptoms that are virtually indistinguishable from those seen in schizophrenia. | Kegeles et al. (2000) [41] | Amphetamine-induced hyperdopaminergic activity in schizophrenia result from disturbances in the glutamatergic neuronal systems that regulate dopaminergic cellular activity. |
| Lahti et al. (1995) [46] Kapur et Seeman (2002) [47] Frohlich and Van Horn (2014) [48] Javitt (2002) [49] | Phencyclidine and ketamine which are dissociative anesthetics act as glutamate antagonists by blocking the glutamate receptor of the N-methyl-D-aspartate (NMDA) type and induce positive, negative, and cognitive symptoms. | ||
| Olney and Farber (1995) [50] Grace (1991) [51] | Impaired dopaminergic neurotransmission in schizophrenia may itself be secondary to the abnormal NMDA-receptor neurotransmission. | ||
| Goldman-Rakic and Selemon (1997) [52] Weinberger and Berman (1996) [53] Du et al. (2019) [54] Walton et al. (2018) [55] | Abnormalities of glutamatergic afferent neurons from the prefrontal cortex to the dopaminergic subcortical areas of the midbrain are associated with abnormal dopamine regulation. | ||
| Coyle (1996) [66] Lisman et al. (2008) [67] | NMDA-receptor deficiency may be the primary element in a dysfunctional brain network leading to dopamine-mediated psychosis in consequence. | ||
| Stone et al. (2007) [85] | Ketamine anesthesia does not cause psychotic symptoms in prepubertal children when compared to anesthesia in adults (onset of schizophrenia in early adulthood). | ||
| Olney et al. (1999) [86] | A chain of neural connections involved in processes generating psychotic phenomena and neurotoxicity result from NMDA-receptor antagonism, and this chain does not fully develop until the end of adolescence (onset of schizophrenia). | ||
| Dysconnection hypothesis | Schizophrenia can be described as impaired connectivity disorder caused by a failure of functional integration in the brain and is based on a model of functional (synaptic) connectivity, specifically an abnormal regulation of synaptic efficacy. | Robinson and Becker (1986) [56] Schmidt and Beninger (2006) [57] | Incentive learning is thought to underpin psychostimulant-induced context-dependent sensitization, which may be important in the development of addiction, dyskinesia, and amphetamine-induced psychosis. |
| Bolton et al. (2020) [72] | The “triple network” system is malfunctioning in schizophrenia. | ||
| Williamson (2007) [84] | Impaired synchronization between the anti-correlated Default mode network and Central executive network is a key pathophysiological feature of schizophrenia. |
| Magnetic-Resonance Imaging Technique | Study | Main Findings/Conclusion |
|---|---|---|
| Structural neuroimaging | Zhuo et al. (2017) [95] | Schizophrenic patients have reduced gray matter volumes in the frontal, temporal, and parietal areas, the cingulate gyrus, and limbic structures (hippocampus, parahippocampus and thalamus). |
| van Erp et al. (2016) [96] | Decrease in the amygdala and increase in the pallidum, which is directly associated to the longevity of the disorder which can be interpreted as evidence in favor of the neurodegenerative hypothesis of schizophrenia. | |
| Chang et al. (2016) [97] | Reduction in grey matter volumes in the vermis, superior temporal gyrus, operculum. | |
| Wright et al. (2000) [98] | Elevated cerebro-spinal fluid volume. | |
| Stoyanov et al. (2021) [99] | Regions located in the left and right opercular part of inferior frontal gyrus, right supramarginal gyrus, left superior temporal gyrus, left anterior orbital gyrus, supplementary motor cortex, and several occipital areas are highly discriminatory for convergent cross-validation of biological features of psychosis vs. depression. | |
| Shepherd et al. (2012) [100] Goodkind et al. (2015) [101] Sheffield et al. (2020) [102] Lee et al. (2016) [103] Mier et al. (2014) [104] | Grey matter volume of the bilateral insula is reduced in psychotic disorders, and a progressive structural decrease is recorded throughout the course and chronicity of the condition. | |
| Functional task-related neuroimaging | Mier et al. (2014) [104] Goghari et al. (2017) [105] Belge et al. (2017) [106] | Reduced accuracy in recognizing emotions and prolonged response time in patients with schizophrenia. |
| Whitfield-Gabrieli et al. (2009) [107] | Task-related hyperactivation in the components of the Default mode network psychotic in patients, while in healthy controls–deactivation in the same network. | |
| Stoyanov et al. (2021) [108] | Activations in Default mode network components during cognitive processing of paranoid-specific items from the von Zerssen’s Paranoid-depressive scale [108]. | |
| Functional resting-state neuroimaging | Nekovarova et al. (2014) [109] | Impaired coordination of Default mode network / Central executive network / Salience network is associated with disorientation between internally and externally focused attention and cognitive impairment. |
| Zhou et al. (2016) [110] | Reduced task-related Default mode network suppression is a psychosis-specific biomarker for cognitive impairment, as the finding is established only in psychotic individuals with cognitive decline. | |
| Stoyanov et al. (2021) [108] Aryutova et al. (2021) [111] | There is a strong aberrant brain connectivity in schizophrenia–an inhibitory influence from prefrontal cortex to Salience network (anterior insula) and an excitatory connection from the anterior cingulate cortex to anterior insula [108,111]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aryutova, K.; Stoyanov, D. Pharmaco-Magnetic Resonance as a Tool for Monitoring the Medication-Related Effects in the Brain May Provide Potential Biomarkers for Psychotic Disorders. Int. J. Mol. Sci. 2021, 22, 9309. https://doi.org/10.3390/ijms22179309
Aryutova K, Stoyanov D. Pharmaco-Magnetic Resonance as a Tool for Monitoring the Medication-Related Effects in the Brain May Provide Potential Biomarkers for Psychotic Disorders. International Journal of Molecular Sciences. 2021; 22(17):9309. https://doi.org/10.3390/ijms22179309
Chicago/Turabian StyleAryutova, Katrin, and Drozdstoy Stoyanov. 2021. "Pharmaco-Magnetic Resonance as a Tool for Monitoring the Medication-Related Effects in the Brain May Provide Potential Biomarkers for Psychotic Disorders" International Journal of Molecular Sciences 22, no. 17: 9309. https://doi.org/10.3390/ijms22179309
APA StyleAryutova, K., & Stoyanov, D. (2021). Pharmaco-Magnetic Resonance as a Tool for Monitoring the Medication-Related Effects in the Brain May Provide Potential Biomarkers for Psychotic Disorders. International Journal of Molecular Sciences, 22(17), 9309. https://doi.org/10.3390/ijms22179309