
Osteoblast Dysfunction in Non-Hereditary Sclerosing Bone Diseases

 ,
,  ,
, 
Abstract
:1. Introduction
2. Osteoblast Physiology
3. Melorheostosis
4. Intramedullary Osteosclerosis
5. Osteoblastic Metastasis
6. Myelofibrosis
7. Sickle Cell Disease
8. Osteoarthritis
9. Erdheim-Chester Disease
10. Paget Disease of the Bone
11. Conclusions
Funding
Conflicts of Interest
References
- Hayashi, M.; Ono, T.; Nakashima, T. Signaling in Osteoblast Differentiation, Zaidi MBT-E. of BB, ed.; Academic Press: Oxford, UK, 2020; pp. 416–426. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugamori, Y.; Mise-Omata, S.; Maeda, C.; Aoki, S.; Tabata, Y.; Murali, R.; Yasuda, H.; Udagawa, N.; Suzuki, H.; Honma, M.; et al. Peptide drugs accelerate BMP-2-induced calvarial bone regeneration and stimulate osteoblast differentiation through mTORC1 signaling. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef]
- Suzuki, A.; Shinoda, J.; Kanda, S.; Oiso, Y.; Kozawa, O. Basic fibroblast growth factor stimulates phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like cells. J. Cell. Biochem. 1996, 63, 491–499. [Google Scholar] [CrossRef]
- Debiais, F.; Lefèvre, G.; Lemonnier, J.; Le Mée, S.; Lasmoles, F.; Mascarelli, F.; Marie, P.J. Fibroblast growth factor-2 induces osteoblast survival through a phosphatidylinositol 3-kinase-dependent, -β-catenin-independent signaling pathway. Exp. Cell Res. 2004, 297, 235–246. [Google Scholar] [CrossRef]
- Mehrotra, M.; Krane, S.M.; Walters, K.; Pilbeam, C. Differential regulation of platelet-derived growth factor stimulated migration and proliferation in osteoblastic cells. J. Cell. Biochem. 2004, 93, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, L.R.; Avioli, L.V. Activation of extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2) by FGF-2 and PDGF-BB in normal human osteoblastic and bone marrow stromal cells: Differences in mobility and in-gel renaturation of ERK1 in human, rat, and mouse osteoblasticce. Biochem. Biophys. Res. Commun. 1997, 238, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Kyono, A.; Avishai, N.; Ouyang, Z.; Landreth, G.E.; Murakami, S. FGF and ERK signaling coordinately regulate mineralization-related genes and play essential roles in osteocyte differentiation. J. Bone Miner. Metab. 2012, 30, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schett, G. Biology, physiology, and morphology of bone. In Kelley and Firestein’s Textbook of Rheumatology; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., Ninth, E., Saunders, W.B., Eds.; O’Dell JRBT-KT of R: Philadelphia, PA, USA, 2013; pp. 61–66. [Google Scholar] [CrossRef]
- Corrado, A.; Sanpaolo, E.R.; Di Bello, S.; Cantatore, F.P. Osteoblast as a target of anti-osteoporotic treatment. Postgrad. Med. 2017, 129, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, A.; Clarke, B.L. Melorheostosis: A Rare Sclerosing Bone Dysplasia. Curr. Osteoporos. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mariaud-Schmidt, R.P.; Emilio Bitar, W.; Pérez-Lamero, F.; Barros-Nuñez, P. Melorheostosis: Unusual presentation in a girl. Clin. Imaging 2002. [Google Scholar] [CrossRef]
- Chou, S.-H.; Chen, C.-H.; Chen, J.-C.; Chien, S.-H.; Cheng, Y.-M. Surgical treatment of melorheostosis: Report of two cases. Kaohsiung J. Med. Sci. 2012, 28, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Kaji, H.; Naito, J.; Sowa, H.; Sugimoto, T.; Chihara, K. Smad3 differently affects osteoblast differentiation depending upon its differentiation stage. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2006, 38, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Jha, S.; Ivovic, A.; Fratzl-Zelman, N.; Deng, Z.; Mitra, A.; Cabral, W.A.; Hanson, E.P.; Lange, E.; Cowen, E.W.; et al. Somatic SMAD3-activating mutations cause melorheostosis by up-regulating the TGF-β/SMAD pathway. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Kang, H.; Jha, S.; Deng, Z.; Fratzl-Zelman, N.; Cabral, W.A.; Ivovic, A.; Meylan, F.; Hanson, E.P.; Lange, E.; Katz, J.; et al. Somatic activating mutations in MAP2K1 cause melorheostosis. Nat. Commun. 2018, 9, 1390. [Google Scholar] [CrossRef] [Green Version]
- Chanchairujira, K.; Chung, C.B.; Lai, Y.M.; Haghighi, P.; Resnick, D. Intramedullary Osteosclerosis: Imaging Features in Nine Patients. Radiology 2001, 220, 225–230. [Google Scholar] [CrossRef]
- Roudier, M.P.; Morrissey, C.; True, L.D.; Higano, C.S.; Vessella, R.L.; Ott, S.M. Histopathological assessment of prostate cancer bone osteoblastic metastases. J. Urol. 2008, 180, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Cheng, C.J.; Bilen, M.A.; Lu, J.F.; Satcher, R.L.; Yu-Lee, L.Y.; Gallick, G.E.; Maity, S.N.; Lin, S.H. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res. 2011, 71, 5194–5203. [Google Scholar] [CrossRef] [Green Version]
- Zhi, G.L.; Mathew, P.; Yang, J.; Starbuck, M.W.; Zurita, A.J.; Liu, J.; Sikes, C.; Multani, A.S.; Efstathiou, E.; Lopez, A.; et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Investig. 2008, 118, 2697–2710. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Gajdosik, M.S.; Josic, D.; Clifton, J.G.; Logothetis, C.; Yu-Lee, L.Y.; Gallick, G.E.; Maity, S.N.; Lin, S.H. Secretome analysis of an osteogenic prostate tumor identifies complex signaling networks mediating cross-talk of cancer and stromal cells within the tumor microenvironment. Mol. Cell. Proteom. MCP 2015, 14, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.J.; Xiong, X.; Ji, X.; Mönkkönen, J.; Russell, R.G.G.; Williamson, M.P.; Ebetino, F.H.; Watts, D.J. Inhibition of growth of Dictyostelium discoideum amoebae by bisphosphonate drugs is dependent on cellular uptake. Pharm. Res. 1997, 14, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Vinholes, J.J.; Purohit, O.P.; Abbey, M.E.; Eastell, R.; Coleman, R.E. Relationships between biochemical and symptomatic response in a double-blind randomised trial of pamidronate for metastatic bone disease. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1997, 8, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Corrado, A.; Gaudio, A.; Ruggieri, S.; Ribatti, D. In vitro and in vivo angiogenic activity of osteoarthritic and osteoporotic osteoblasts is modulated by VEGF and vitamin D3 treatment. Regul. Pept. 2013, 184, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Rameshwar, P.; Narayanan, R.; Qian, J.; Denny, T.N.; Colon, C.; Gascon, P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: An inflammatory response beyond the realm of homeostasis. J. Immunol. 2000, 165, 2271–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Bianchi, L.; Paoletti, F.; Pancrazzi, A.; Torre, E.; Nishikawa, M.; Zingariello, M.; Di Baldassarre, A.; Rana, R.A.; Lorenzini, R.; et al. A pathobiologic pathway linking thrombopoietin, GATA-1, and TGF-beta1 in the development of myelofibrosis. Blood 2005, 105, 3493–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.P.; Hunter, D.J. Managing osteoarthritis. Aust. Prescr. 2015, 38, 115–119. Available online: https://pubmed.ncbi.nlm.nih.gov/26648637 (accessed on 10 June 2021). [CrossRef] [PubMed] [Green Version]
- Lourido, L.; Calamia, V.; Mateos, J.; Fernández-Puente, P.; Fernández-Tajes, J.; Blanco, F.J.; Ruiz-Romero, C. Quantitative proteomic profiling of human articular cartilage degradation in osteoarthritis. J. Proteome Res. 2014, 13, 6096–6106. [Google Scholar] [CrossRef]
- Tat, S.K.; Padrines, M.; Theoleyre, S.; Couillaud-Battaglia, S.; Heymann, D.; Redini, F.; Fortun, Y. OPG/membranous--RANKL complex is internalized via the clathrin pathway before a lysosomal and a proteasomal degradation. Bone 2006, 39, 706–715. [Google Scholar] [CrossRef]
- Tat, S.K.; Pelletier, J.-P.; Lajeunesse, D.; Fahmi, H.; Duval, N.; Martel-Pelletier, J. Differential modulation of RANKL isoforms by human osteoarthritic subchondral bone osteoblasts: Influence of osteotropic factors. Bone 2008, 43, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Higuchi, T.; Taniguchi, Y.; Yonezawa, H.; Araki, Y.; et al. Diagnosis and treatment of intramedullary osteosclerosis: A report of three cases and literature review. BMC Musculoskelet. Disord. 2020, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Dawson, N. How is androgen-dependent metastatic prostate cancer best treated? Hematol. Oncol. Clin. N. Am. 1996, 10, 727–747. [Google Scholar] [CrossRef]
- Coleman, R.E. Skeletal complications of ma lignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Choueiri, M.B.; Tu, S.-M.; Yu-Lee, L.-Y.; Lin, S.-H. The central role of osteoblasts in the metastasis of prostate cancer. Cancer Metastasis Rev. 2006, 25, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; Debiais, F.; Haÿ, E. Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2 and BMP-2 signaling. Histol. Histopathol. 2002, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Decker, K.F.; Zhou, T.; Chen, J.; Qi, Z.; Jacobs, K.; Weilbaecher, K.N.; Corey, E.; Long, F.; Jia, L. Role of WNT7B-induced noncanonical pathway in advanced prostate cancer. Mol. Cancer Res. 2013, 11, 482–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaeminck-Guillem, V. Extracellular Vesicles in Prostate Cancer Carcinogenesis, Diagnosis, and Management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef]
- Scimeca, M.; Urbano, N.; Rita, B.; Mapelli, S.N.; Catapano, C.V.; Carbone, G.M.; Ciuffa, S.; Tavolozza, M.; Schillaci, O.; Mauriello, A.; et al. Prostate Osteoblast-Like Cells: A Reliable Prognostic Marker of Bone Metastasis in Prostate Cancer Patients. Contrast Media Mol. Imaging 2018, 2018, 9840962. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 2017, 8, 94834–94849. [Google Scholar] [CrossRef] [Green Version]
- Clézardin, P. Pathophysiology of bone metastases from solid malignancies. Jt. Bone Spine 2017, 84, 677–684. [Google Scholar] [CrossRef]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef]
- Dedes, P.G.; Gialeli, C.; Tsonis, A.I.; Kanakis, I.; Theocharis, A.D.; Kletsas, D.; Tzanakakis, G.N.; Karamanos, N.K. Expression of matrix macromolecules and functional properties of breast cancer cells are modulated by the bisphosphonate zoledronic acid. Biochim. Biophys. Acta 2012, 1820, 1926–1939. [Google Scholar] [CrossRef]
- Huang, W.-W.; Huang, C.; Liu, J.; Zheng, H.-Y.; Lin, L. Zoledronic acid as an adjuvant therapy in patients with breast cancer: A systematic review and meta-analysis. PLoS ONE 2012, 7, e40783. [Google Scholar] [CrossRef] [Green Version]
- Chandran, T.; Venkatachalam, I. Efficacy and safety of denosumab compared to bisphosphonates in improving bone strength in postmenopausal osteoporosis: A systematic review. Singap. Med. J. 2019, 60, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Loriot, Y.; McGregor, B.A.; Dreicer, R.; Dorff, T.B.; Maughan, B.L.; Kelly, W.K.; Pagliaro, L.C.; Srinivas, S.; Squillante, C.M.; et al. Cabozantinib in combination with atezolizumab in patients with metastatic castration-resistant prostate cancer: Results of cohort 6 of the COSMIC-021 study. J. Clin. Oncol. 2020, 38, 5564. [Google Scholar] [CrossRef]
- Psaila, B.; Wang, G.; Rodriguez-Meira, A.; Li, R.; Heuston, E.F.; Murphy, L.; Yee, D.; Hitchcock, I.S.; Sousos, N.; O’Sullivan, J.; et al. Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets. Mol. Cell 2020, 78, 477–492.e8. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Litzow, M.R.; Pardanani, A. Long-Term Outcome of Treatment with Ruxolitinib in Myelofibrosis. N. Engl. J. Med. 2011, 365, 1455–1457. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.P.; Block, M.H. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with the myeloproliferative syndrome. Medicine 1971, 50, 357–420. [Google Scholar] [CrossRef]
- Curto-Garcia, N.; Harrison, C.; McLornan, D.P. Bone marrow niche dysregulation in myeloproliferative neoplasms. Haematologica 2020, 105, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Hemavathy, K.; Charles, W.; Zhang, H.; Dua, P.K.; Novetsky, A.D.; Chang, T.; Wong, C.; Jabara, M. Osteosclerosis in idiopathic myelofibrosis is related to the overproduction of osteoprotegerin (OPG). Exp. Hematol. 2004, 32, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Le Bousse-Kerdilès, M.C.; Chevillard, S.; Charpentier, A.; Romquin, N.; Clay, D.; Smadja-Joffe, F.; Praloran, V.; Dupriez, B.; Demory, J.L.; Jasmin, C.; et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in CD34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood 1996, 88, 4534–4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Malaspina, H. Pathogenesis of myelofibrosis: Role of ineffective megakaryopoiesis and megakaryocyte components. Prog. Clin. Biol. Res. 1984, 154, 427–454. [Google Scholar] [PubMed]
- Zhang, J.; Fu, M.; Myles, D.; Zhu, X.; Du, J.; Cao, X.; Chen, Y.E. PDGF induces osteoprotegerin expression in vascular smooth muscle cells by multiple signal pathways. FEBS Lett. 2002, 521, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Stoupa, E.; Antoniadou, L.; Premetis, E.; Konstantopoulos, K.; Papassotiriou, I.; Terpos, E. Osteoporosis and osteosclerosis in sickle cell/beta-thalassemia: The role of the RANKL/osteoprotegerin axis. Haematologica 2006, 91, 813–816. [Google Scholar]
- Mokhtar, G.M.; Tantawy, A.A.G.; Hamed, A.A.-S.; Adly, A.A.M.; Ismail, E.A.R.; Makkeyah, S.M. Tartrate-Resistant Acid Phosphatase 5b in Young Patients with Sickle Cell Disease and Trait Siblings: Relation to Vasculopathy and Bone Mineral Density. Clin. Appl. Thromb. Hemost. 2015, 23, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Rana, K.; Pantoja, K.; Xiao, L. Bone marrow neutrophil aging in sickle cell disease mice is associated with impaired osteoblast functions. Biochem. Biophys. Rep. 2018, 16, 110–114. [Google Scholar] [CrossRef]
- Ware, R.E.; Aygun, B. Advances in the use of hydroxyurea. Hematology 2009, 2009, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Suri, S.; Walsh, D. Osteochondral alterations in osteoarthritis. Bone 2011, 51, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jordan, J.M. Epidemiology of osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, A.; Maruotti, N.; Cantatore, F.P. Osteoblast Role in Rheumatic Diseases. Int. J. Mol. Sci. 2017, 18, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Wan, F.; Zhou, Z.; Tao, R.; Lu, Y.; Zhou, M.; Liu, F.; Liu, Y. Identification of key regulators responsible for dysregulated networks in osteoarthritis by large-scale expression analysis. J. Orthop. Surg. Res. 2021, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Attur, M.; Yang, Q.; Shimada, K.; Tachida, Y.; Nagase, H.; Mignatti, P.; Statman, L.; Palmer, G.; Kirsch, T.; Beier, F.; et al. Elevated expression of periostin in human osteoarthritic cartilage and its potential role in matrix degradation via matrix metalloproteinase-13. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4107–4121. [Google Scholar] [CrossRef] [Green Version]
- Attur, M.; Duan, X.; Cai, L.; Han, T.; Zhang, W.; Tycksen, E.D.; Samuels, J.; Brophy, R.H.; Abramson, S.B.; Rai, M.F. Periostin loss-of-function protects mice from post-traumatic and age-related osteoarthritis. Arthritis Res. Ther. 2021, 23, 104. [Google Scholar] [CrossRef]
- Bettica, P.; Cline, G.; Hart, D.J.; Meyer, J.; Spector, T.D. Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheum. 2002, 46, 3178–3184. [Google Scholar] [CrossRef]
- Klose-Jensen, R.; Hartlev, L.B.; Boel, L.W.T.; Laursen, M.B.; Stengaard-Pedersen, K.; Keller, K.K.; Hauge, E.M. Subchondral bone turnover, but not bone volume, is increased in early stage osteoarthritic lesions in the human hip joint. Osteoarthr. Cartil. 2015, 23, 2167–2173. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Chan, P.M.B.; Wen, C. Do immune cells lead the way in subchondral bone disturbance in osteoarthritis? Prog. Biophys. Mol. Biol. 2019, 148, 21–31. [Google Scholar] [CrossRef]
- Netzer, C.; Urech, K.; Hügle, T.; Benz, R.M.; Geurts, J.; Schären, S. Characterization of subchondral bone histopathology of facet joint osteoarthritis in lumbar spinal stenosis. J. Orthop. Res. 2016, 34, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Corrado, A.; Neve, A.; Macchiarola, A.; Gaudio, A.; Marucci, A.; Cantatore, F.P. RANKL/OPG ratio and DKK-1 expression in primary osteoblastic cultures from osteoarthritic and osteoporotic subjects. J. Rheumatol. 2013, 40, 684–694. [Google Scholar] [CrossRef]
- Chester, W. Über Lipoidgranulomatose. Virchows Arch. Für Pathol. Anat. Phys. Für Klin. Med. 1930, 279, 561–602. [Google Scholar] [CrossRef]
- Estrada-Veras, J.I.; O’Brien, K.J.; Boyd, L.C.; Dave, R.H.; Durham, B.H.; Xi, L.; Malayeri, A.A.; Chen, M.Y.; Gardner, P.J.; Alvarado Enriquez, J.R.; et al. The clinical spectrum of Erdheim-Chester disease: An observational cohort study. Blood Adv. 2017, 1, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Haroche, J.; Arnaud, L.; Cohen-Aubart, F.; Hervier, B.; Charlotte, F.; Emile, J.F.; Amoura, Z. Erdheim–Chester Disease. Curr. Rheumatol. Rep. 2014, 16, 412. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-Z.; Zhou, X.; Song, A.; Wang, Y.-P.; Liu, Y. Exophthalmos and coated aorta in Erdheim–Chester disease. Rheumatology 2020, 59, 2651–2652. [Google Scholar] [CrossRef]
- Ben-Yaakov, G.; Munteanu, D.; Sztarkier, I.; Fich, A.; Schwartz, D. Erdheim Chester—A rare disease with unique endoscopic features. World J. Gastroenterol. 2014, 20, 8309–8311. [Google Scholar] [CrossRef]
- Veyssier-Belot, C.; Cacoub, P.; Caparros-Lefebvre, D.; Wechsler, J.; Brun, B.; Remy, M.; Wallaert, B.; Petit, H.; Grimaldi, A.; Wechsler, B.; et al. Erdheim-Chester Disease Clinical and Radiologic Characteristics of 59 Cases. Medicine 1996, 75, 157–169. Available online: https://journals.lww.com/md-journal/Fulltext/1996/05000/Erdheim_Chester_Disease_Clinical_and_Radiologic.5.aspx (accessed on 9 June 2021). [CrossRef]
- Haroche, J.; Papo, M.; Cohen-Aubart, F.; Charlotte, F.; Maksud, P.; Grenier, P.A.; Cluzel, P.; Mathian, A.; Emile, J.F.; Amoura, Z. La maladie d’Erdheim-Chester, une néoplasie myéloïde inflammatoire. La Presse Med. 2017, 46, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Heaney, M.L.; Collin, M.; Cohen-Aubart, F.; Vaglio, A.; Durham, B.H.; Hershkovitz-Rokah, O.; Girschikofsky, M.; Jacobsen, E.D.; Toyama, K.; et al. Erdheim-Chester disease: Consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020, 135, 1929–1945. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Durham, B.H.; Haroche, J.; Yao, Z.; Ma, J.; Parikh, S.A.; Wang, Z.; Choi, J.; Kim, E.; Cohen-Aubart, F.; et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016, 6, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.; Ralston, S.H. Clinical presentation of Paget’s disease: Evaluation of a contemporary cohort and systematic review. Calcif. Tissue Int. 2014, 95, 385–392. [Google Scholar] [CrossRef]
- Hosking, D.J. Paget’s disease of bone. Br. Med. J. Clin. Res. Ed. 1981, 283, 686–688. [Google Scholar] [CrossRef] [Green Version]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Miyagawa, K.; Ohata, Y.; Petrusca, D.N.; Pagnotti, G.M.; Mohammad, K.S.; Guise, T.A.; Windle, J.J.; David Roodman, G.; Kurihara, N. Increased S1P expression in osteoclasts enhances bone formation in an animal model of Paget’s disease. J. Cell. Biochem. 2021, 122, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.V. Etiology of Paget’s disease and osteoclast abnormalities. J. Cell. Biochem. 2004, 93, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Kravets, I. Paget’s Disease of Bone: Diagnosis and Treatment. Am. J. Med. 2018, 131, 1298–1303. [Google Scholar] [CrossRef]


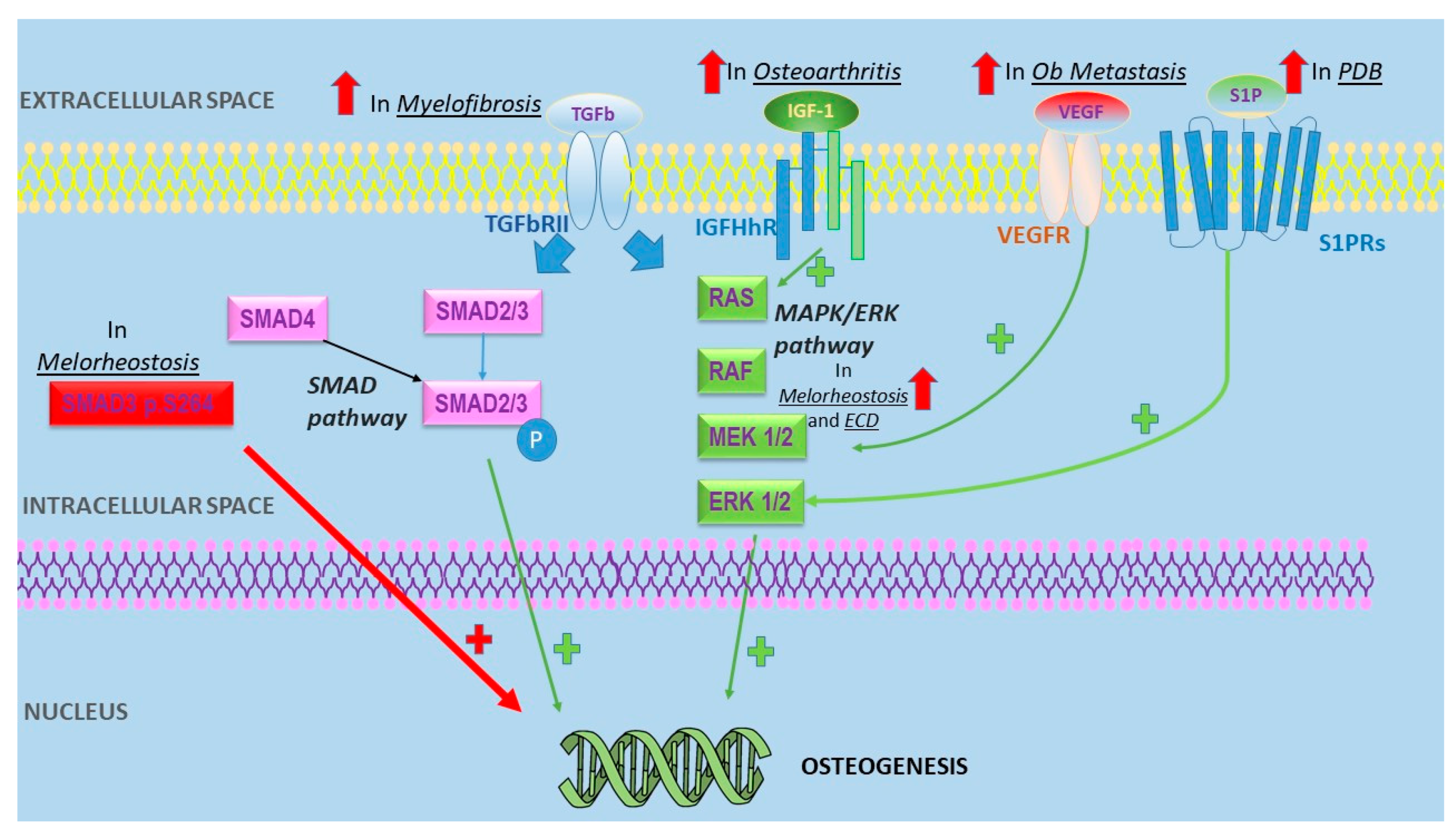{kind=link}
| Disease | Pathogenic Mechanisms Leading to Osteosclerosis | References |
|---|---|---|
| Melorheostosis | -Mutations of MAP2K1 encoding MEK1-ERK1/2 proteins -SMAD3 p.S264 substitutions were shown to increase TGF-β signaling | [16,17] |
| Osteoblastic metastases | -MDA-BF-1 expressed only in PCa bone metatasis -BMP paracrine effect on PCa -Cross-talk between PCa and osteoblasts -Osteomimicry -Micro-RNA transferring from PCa to osteoblasts -Altered VEGF expression | [19,20,21,22,23,24] |
| Myelofibrosis | -Correlation between bone sclerosis and OPG serum level -High FGF and TGF levels | [25,26] |
| Sickle cell disease | -Increased activity after osteoblast inhibition by iron overload -RANK/RANKL/OPG and Wnt/β-catenin pathways alterations | [27,28] |
| Osteoarthritis | -JUN disregulation -Periostin upregulation -IGF-1 expression may promote bone sclerosis | [29,30,31] |
| Erdheim Chester disease | Activation of MAPK pathway | [32] |
| Paget disease of the bone | -Sphingosine-1-phosphate (S1P) overexpression leads to osteoblasts hyperactivity -MV mRNA commonly observed in Pagetic osteoclasts | [33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giardullo, L.; Altomare, A.; Rotondo, C.; Corrado, A.; Cantatore, F.P. Osteoblast Dysfunction in Non-Hereditary Sclerosing Bone Diseases. Int. J. Mol. Sci. 2021, 22, 7980. https://doi.org/10.3390/ijms22157980
Giardullo L, Altomare A, Rotondo C, Corrado A, Cantatore FP. Osteoblast Dysfunction in Non-Hereditary Sclerosing Bone Diseases. International Journal of Molecular Sciences. 2021; 22(15):7980. https://doi.org/10.3390/ijms22157980
Chicago/Turabian StyleGiardullo, Liberato, Alberto Altomare, Cinzia Rotondo, Addolorata Corrado, and Francesco Paolo Cantatore. 2021. "Osteoblast Dysfunction in Non-Hereditary Sclerosing Bone Diseases" International Journal of Molecular Sciences 22, no. 15: 7980. https://doi.org/10.3390/ijms22157980
APA StyleGiardullo, L., Altomare, A., Rotondo, C., Corrado, A., & Cantatore, F. P. (2021). Osteoblast Dysfunction in Non-Hereditary Sclerosing Bone Diseases. International Journal of Molecular Sciences, 22(15), 7980. https://doi.org/10.3390/ijms22157980