
Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glucocorticoids and Hypoxia-Brain Tolerance and Cross-Talk Mechanisms
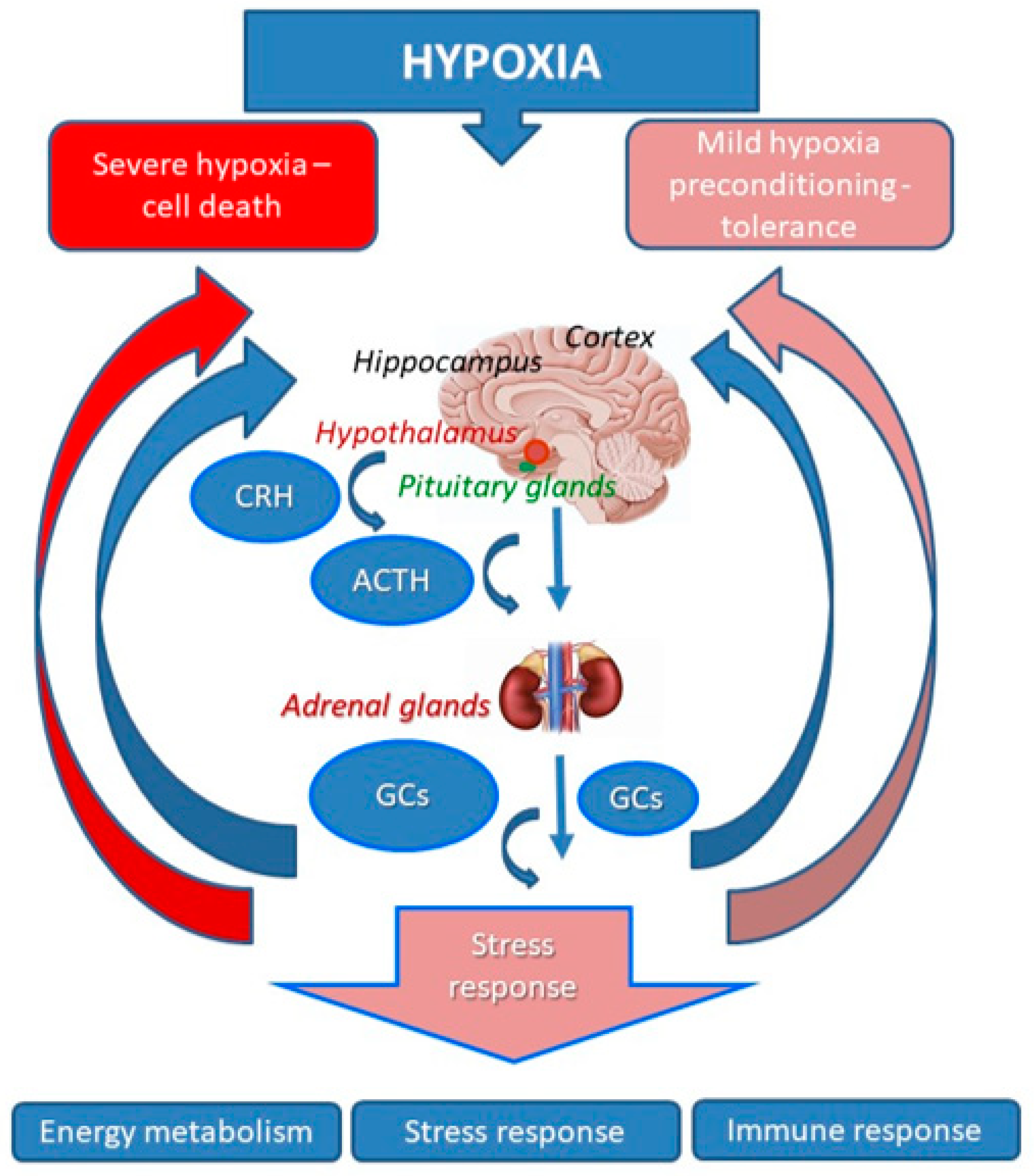
2.1. Dynamics of the HPA Activation in Injurious and Non-Injurious Hypoxia
2.2. Glucocorticoid Receptors of the Brain
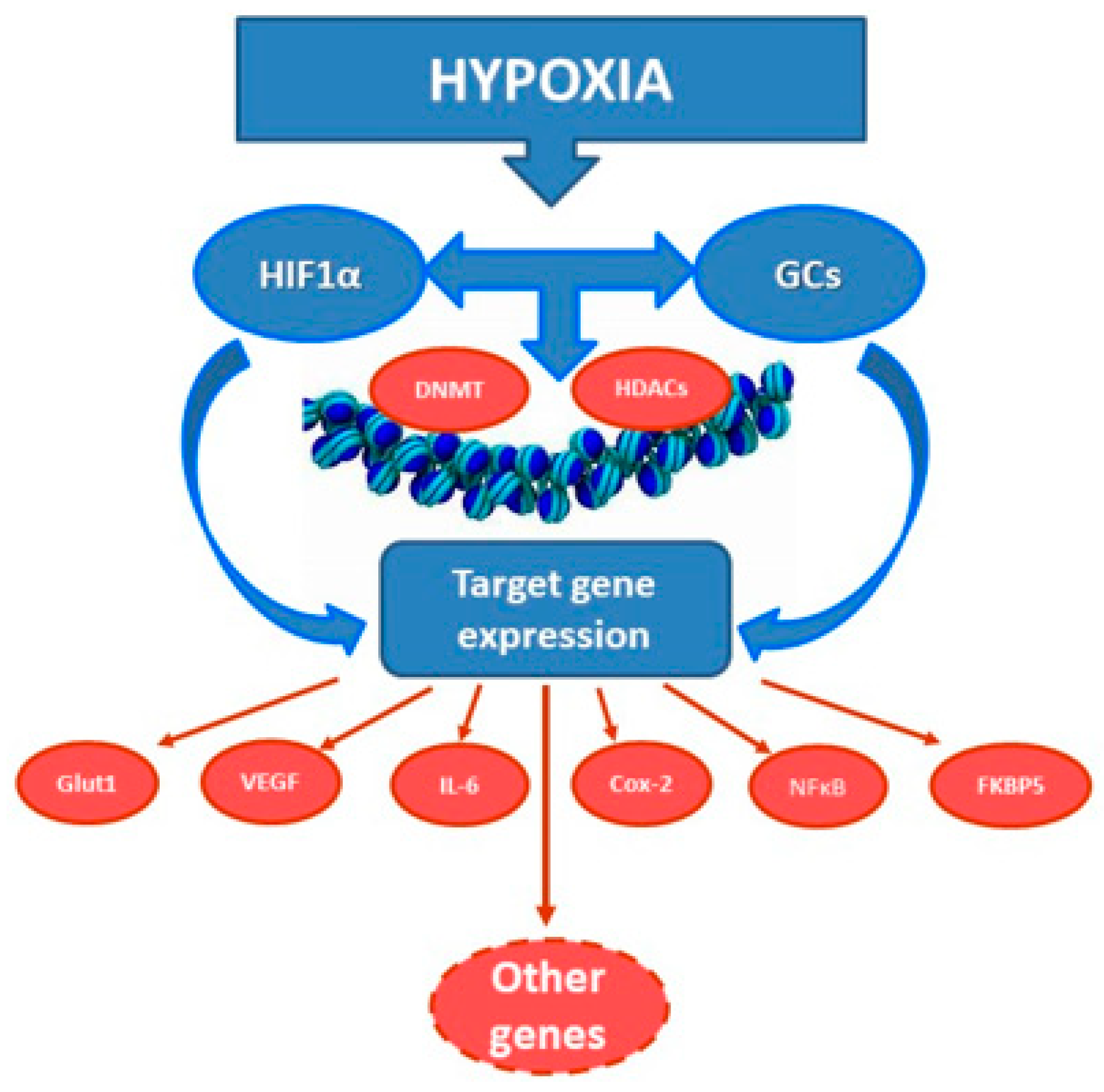
2.3. Interaction of GCs and Hypoxia-Inducible Factor HIF-1
2.4. A Cross-Talk between Glucocorticoid and Hypoxic Signaling
2.5. Glucocorticoids, Hypoxia and Inflammation
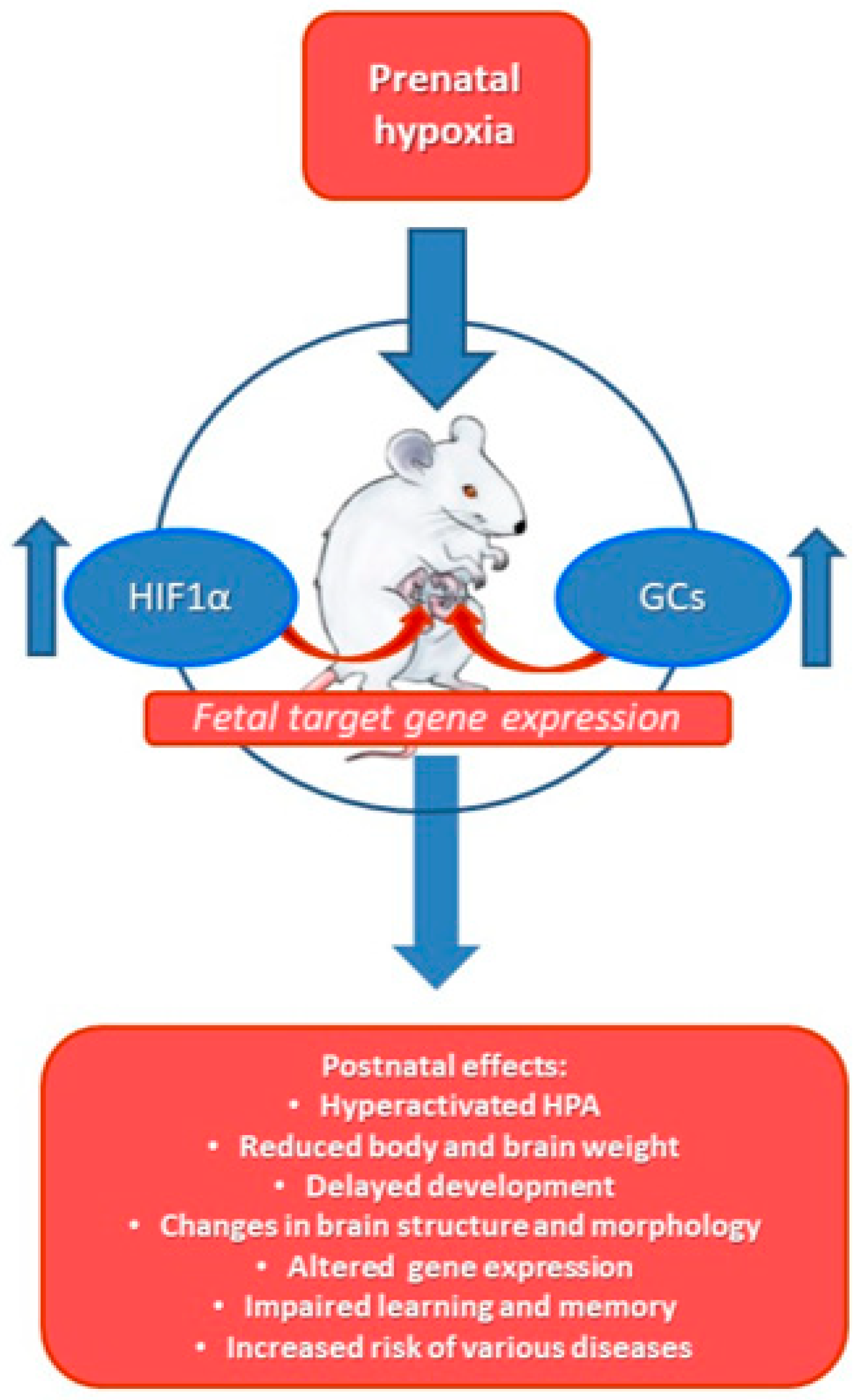
2.6. Effects of Prenatal Hypoxia on HPA, GC and Hypoxic Response in Later Life
3. Summary
4. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- McEwen, B.S. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [Green Version]
- Aquilera, G. HPA axis responsiveness to stress: Implications for healthy aging. Exp. Gerontol. 2011, 46, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Kellendonk, C.; Gass, P.; Kretz, O.; Schutz, G.; Tronche, F. Corticosteroid receptors in the brain: Gene targeting studies. Brain Res. Bull. 2002, 57, 73–83. [Google Scholar] [CrossRef]
- Rybnikova, E.A.; Samoilov, M.O. Current insights into the molecular mechanisms of hypoxic pre- and postconditioning using hypobaric hypoxia. Review. Front. Neurosci. 2015, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hafeez, F.; Noorulla, F.; Geng, X.; Shao, G.; Ren, C.; Lu, G.; Zhao, H.; Ding, Y.; Ji, X. Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 2017, 157, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Tuor, U.I.; Del Bigio, M.R.; Chumas, P.D. Brain damage due to cerebral hypoxia/ischemia in the neonate: Pathology and pharmacological modification. Cerebrovasc. Brain Metab. Rev. 1996, 8, 159–193. [Google Scholar]
- Kaljusto, M.-L.; Stensløkken, K.-O.; Mori, T.; Panchenko, A.; Frantzen, M.-L.; Valen, G.; Vaage, J. Preconditioning effects of steroids and hyperoxia on cardiac ischemia-reperfusion injury and vascular reactivity. Eur. J. Cardiothorac. Surg. 2008, 33, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobryshev, P.; Bagaeva, T.; Filaretova, L. Ischemic preconditioning attenuates gastric ischemia-reperfusion injury through involvement of glucocorticoids. J. Physiol. Pharmacol. 2009, 60 (Suppl. S7), 155–160. [Google Scholar] [PubMed]
- Rybnikova, E.A.; Mironova, V.I.; Pivina, S.G.; Ordyan, N.E.; Tulkova, E.I.; Samoilov, M.O. Hormonal mechanisms of neuroprotective effects of the mild hypoxic preconditioning in rats. Dokl. Biol. Sci. 2008, 421, 239–240. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Bhatt, A.J. Corticosteroid responses following hypoxic preconditioning provide neuroprotection against subsequent hypoxic-ischemic brain injury in the newborn rats. Int. J. Dev. Neurosci. 2015, 44, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Glanemann, M.; Strenziok, R.; Kuntze, R.; Münchow, S.; Dikopoulos, N.; Lippek, F.; Langrehr, J.M.; Dietel, M.; Neuhaus, P.; Nussler, A.K. Ischemic preconditioning and methylprednisolone both equally reduce hepatic ischemia/reperfusion injury. Surgery 2004, 135, 203–214. [Google Scholar] [CrossRef]
- Davis, C.; Hackett, P. Advances in the Prevention and Treatment of High Altitude Illness. Emerg. Med. Clin. North. Am. 2017, 35, 241–260. [Google Scholar] [CrossRef]
- Millet, G.P.; Debevec, T.; Brocherie, F.; Burtscher, M.; Burtscher, J. Altitude and COVID-19: Friend or foe? A narrative review. Physiol. Rep. 2021, 8, e14615. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lu, A.; Sharp, F.R. Regional genome transcriptional response of adult mouse brain to hypoxia. BMC Genom. 2011, 12, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerman, A.; Ram, O.; Ifergane, G.; Matar, M.A.; Kaplan, Z.; Hoffman, J.R.; Sadot, O.; Cohen, H. Role of endogenous and exogenous corticosterone on behavioral and cognitive responses to low-pressure blast wave exposure. J. Neurotrauma 2019, 36, 380–394. [Google Scholar] [CrossRef]
- Cernak, I.; Savic, V.J.; Lazarov, A.; Joksimovic, M.; Markovic, S. Neuroendocrine responses following graded traumatic brain injury in male adults. Brain Inj. 1999, 13, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345. [Google Scholar] [CrossRef]
- Weidenfeld, J.; Leker, R.R.; Gai, N.; Teichner, A.; Bener, D.; Ovadia, H. The function of the adrenocortical axis in permanent middle cerebral artery occlusion: Effect of glucocorticoids on the neurological outcome. Brain Res. 2011, 1407, 90–96. [Google Scholar] [CrossRef]
- Rybnikova, E.; Mironova, V.; Pivina, S.; Tulkova, E.; Ordyan, N.; Nalivaeva, N.; Turner, A.; Samoilov, M. Involvement of hypothalamic-pituitary-adrenal axis in the antidepressant-like effects of mild hypoxic preconditioning in rats. Psychoneuroendocrinology 2007, 32, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Vorobyev, M.; Pivina, S.; Samoilov, M. Postconditioning by mild hypoxic exposures reduces rat brain injury caused by severe hypoxia. Neurosci. Lett. 2012, 513, 100–105. [Google Scholar] [CrossRef]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure-function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I. Structural variants of glucocorticoid receptor binding sites and different versions of positive glucocorticoid responsive elements: Analysis of GR-TRRD database. J. Steroid Biochem. Mol. Biol. 2009, 115, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meijsing, S.H. Mechanisms of glucocorticoid-regulated gene transcription. Adv. Exp. Med. Biol. 2015, 872, 59–81. [Google Scholar]
- Merkulov, V.M.; Merkulova, T.I.; Bondar, N.P. Mechanisms of Brain Glucocorticoid Resistance in Stress-Induced Psychopathologies. Biochemistry 2017, 82, 351–365. [Google Scholar] [CrossRef]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef]
- Vedder, H.; Weiss, I.; Holsboer, F.; Reul, J.M. Glucocorticoid and mineralocorticoid receptors in rat neocortical and hippocampal brain cells in culture: Characterization and regulatory studies. Brain Res. 1993, 605, 18–24. [Google Scholar] [CrossRef]
- Podgorny, O.V.; Gulyaeva, N.V. Glucocorticoid-mediated mechanisms of hippocampal damage: Contribution of subgranular neurogenesis. J. Neurochem. 2021, 157, 370–392. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef]
- Dengler, V.; Galbraith, M.; Espinosa, J. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Chavez, J.; Agani, F.; Pichiule, P.; LaManna, J. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J. Appl. Physiol. 2000, 89, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.; Dang, C. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Bragina, O.; Xu, Y.; Cao, Z.; Chen, H.; Zhou, B.; Morgan, M.; Lin, Y.; Jiang, B.; Liu, K.; et al. Glucose up-regulates HIF-1 alpha expression in primary cortical neurons in response to hypoxia through maintaining cellular redox status. J. Neurochem. 2008, 105, 1849–1860. [Google Scholar] [CrossRef]
- Sheldon, R.; Lee, C.; Jiang, X.; Knox, R.; Ferriero, D. Hypoxic preconditioning protection is eliminated in HIF-1α knockout mice subjected to neonatal hypoxia-ischemia. Pediatr. Res. 2014, 76, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukyanova, L.; Kirova, Y. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Brix, B.; Mesters, J.R.; Pellerin, L.; Johren, O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J. Neurosci. 2012, 32, 9727–9735. [Google Scholar] [CrossRef]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 2020, 23, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Segarra-Mondejar, M.; Casellas-Díaz, S.; Ramiro-Pareta, M.; Müller-Sánchez, C.; Martorell-Riera, A.; Hermelo, I.; Reina, M.; Aragonés, J.; Martínez-Estrada, O.M.; Soriano, F.X. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018, 37, e97368. [Google Scholar] [CrossRef]
- Jaszczyk, A.; Juszczak, G.R. Glucocorticoids, metabolism and brain activity. Neurosci. Biobehav. Rev. 2021, 126, 113–145. [Google Scholar] [CrossRef] [PubMed]
- Wacker, B.K.; Perfater, J.L.; Gidday, J.M. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem. 2012, 123, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, C.; Du, X.; Liu, M.; Ji, X.; Du, H.; Zhao, H. Hypoxia Inducible Factor 1alpha Plays a Key Role in Remote Ischemic Preconditioning Against Stroke by Modulating Inflammatory Responses in Rats. J. Am. Heart Assoc. 2018, 7, e007589. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Chang, Y.; Cheng, X.; Luo, X.; Zhang, J.; Tang, X. Postconditioning with repeated mild hypoxia protects neonatal hypoxia-ischemic rats against brain damage and promotes rehabilitation of brain function. Brain Res. Bull. 2018, 139, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Vetrovoy, O.; Sarieva, K.; Galkina, O.; Eschenko, N.; Lyanguzov, A.; Gluschenko, T.; Tyulkova, E.; Rybnikova, E. Neuroprotective mechanism of hypoxic post-conditioning involves HIF1-associated regulation of the pentose phosphate pathway in rat brain. Neurochem. Res. 2019, 44, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Samoilov, M.O.; Sidorova, M.V.; Glushchenko, T.S. The Pattern of the Expression of Hypoxia_Inducible Factor (HIF-1α) in the Neocortex and Hippocampus of Rats after the Presentation of Different Modes of Hypobaric Hypoxia. Neurochem. J. 2015, 9, 299–305. [Google Scholar] [CrossRef]
- Sidorova, M.V.; Rybnikova, E.A.; Churilova, A.V.; Portnichenko, V.I.; Samoilov, M.O. The effect of mild hypobaric hypoxia regime on expression of factor induced by hypoxia in the rat neocortex. Fiziol. Zh. 2013, 59, 111–115. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Zenko, M.Y.; Rybnikova, E.A. Antidepressant-like action of hypoxic postconditioning is accompanied by the up-regulation of hippocampal HIF-1α and erythropoietin. Med. Acad. J. 2019, 19, 41–46. [Google Scholar] [CrossRef]
- Ferlazzo, N.; Currò, M.; Giunta, M.L.; Longo, D.; Rizzo, V.; Caccamo, D.; Ientile, R. Up-regulation of HIF-1alpha is associated with neuroprotective effects of agmatine against rotenone-induced toxicity in differentiated SH-SY5Y cells. Amino Acids 2020, 52, 171–179. [Google Scholar] [CrossRef]
- Li, Y.Q.; Hui, Z.R.; Tao, T.; Shao, K.Y.; Liu, Z.; Li, M.; Gu, L.L. Protective effect of hypoxia inducible factor-1alpha gene therapy using recombinant adenovirus in cerebral ischaemia-reperfusion injuries in rats. Pharm. Biol. 2020, 58, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, C.; Wang, J.; Zhu, M.; Yao, Y.; Liu, J. Hypoxia preconditioning protects neuronal cells against traumatic brain injury through stimulation of glucose transport mediated by HIF-1alpha/GLUTs signaling pathway in rat. Neurosurg. Rev. 2021, 44, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrovoy, O.; Sarieva, K.; Lomert, E.; Nimiritsky, P.; Eschenko, N.; Galkina, O.; Lyanguzov, A.; Tyulkova, E.; Rybnikova, E. Pharmacological HIF1 Inhibition Eliminates Downregulation of the Pentose Phosphate Pathway and Prevents Neuronal Apoptosis in Rat Hippocampus Caused by Severe Hypoxia. J. Mol. Neurosci. 2020, 70, 635–646. [Google Scholar] [CrossRef]
- Kirova, Y.; Shakova, F.; Germanova, E.; Paltsyn, A.G.; Rybnikova, E.A.; Lukyanova, L.D. Urgent changes in the expression of hypoxia-inducible factor-1α (HIF-1α) in the neocortex of rats with different tolerance to acute hypoxia underwent focal ischemic stroke prefrontal cortex. Patol. Fiziol. Eksp. Ter. 2014, 3, 9–16. (In Russian) [Google Scholar]
- Yuan, D.; Guan, S.; Wang, Z.; Ni, H.; Ding, D.; Xu, W.; Li, G. HIF-1alpha aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J. Chem. Neuroanat. 2021, 116, 101994. [Google Scholar] [CrossRef] [PubMed]
- Vangeison, G.; Carr, D.; Federoff, H.J.; Rempe, D.A. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J. Neurosci. 2008, 28, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Shimizu, N.; Yoshikawa, N.; Makino, Y.; Ouchida, R.; Okamoto, K.; Hisada, T.; Nakamura, H.; Morimoto, C.; Tanaka, H. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J. Biol. Chem. 2003, 278, 33384–33391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.E.; Huck, G.; Stiehl, D.P.; Jelkmann, W.; Hellwig-Bürgel, T. Dexamethasone impairs hypoxia-inducible factor-1 function. Biochem. Biophys. Res. Commun. 2008, 372, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Park, C.; Shim, M.K.; Lee, Y.-H.; Lee, Y.M.; Lee, Y. Glucocorticoids suppress hypoxia-induced COX-2 and hypoxia inducible factor-1α expression through the induction of glucocorticoid-induced leucine zipper. Br. J. Pharmacol. 2014, 171, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Vanderhaeghen, T.; Beyaert, R.; Libert, C. Bidirectional Crosstalk Between Hypoxia Inducible Factors and Glucocorticoid Signalling in Health and Disease. Front. Immunol. 2021, 12, 684085. [Google Scholar] [CrossRef]
- Vettori, A.; Greenald, D.; Wilsone, G.K.; Peron, M.; Facchinello, N.; Markham, E.; Sinnakaruppan, M.; Matthews, L.; McKeatingg, J.; Argenton, F.; et al. Glucocorticoids promote Von Hippel Lindau degradation and Hif-1α stabilization. Proc. Nat. Acad. Sci. USA 2017, 114, 9948–9953. [Google Scholar] [CrossRef] [Green Version]
- Baranova, K.A.; Mironova, V.I.; Rybnikova, E.A.; Samoilov, M.O. Characteristics of the transcription factor HIF-1α expression in the rat brain during the development of a depressive state and the antidepressive effects of hypoxic preconditioning. Neurochem. J. 2010, 4, 35–40. [Google Scholar] [CrossRef]
- Marchi, D.; Santhakumar, K.; Markham, E.; Li, N.; Storbeck, K.-H.; Krone, N.; Cunliffe, V.T.; van Eeden, F.J.M. Bidirectional crosstalk between Hypoxia-Inducible Factor and glucocorticoid signaling in zebrafish larvae. PLoS Genet. 2020, 16, e1008757. [Google Scholar] [CrossRef]
- Rybnikova, E.; Glushchenko, T.; Churilova, A.; Pivina, S.; Samoilov, M. Expression of glucocorticoid and mineralocorticoid receptors in hippocampus of rats exposed to various modes of hypobaric hypoxia: Putative role in hypoxic preconditioning. Brain Res. 2011, 1381, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-X.; Pan, X.-Y.; Jin, Y.-D.; Wang, Y.; Song, X.-L.; Wang, C.-H.; Li, Y.-D.; Lu, J. The mechanisms and significance of up-regulation of RhoB expression by hypoxia and glucocorticoid in rat lung and A549 cells. J. Cell. Mol. Med. 2016, 20, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Su, J.; Zhang, M.; Jin, Y.; Wang, Y.; Zhou, P.; Lu, J. RhoB regulates the function of macrophages in the hypoxia-induced inflammatory response. Cell. Mol. Immunol. 2017, 14, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- González, M.V.; Jiménez, B.; Berciano, M.T.; González-Sancho, J.M.; Caelles, C.; Lafarga, M.; Muñoz, A. Glucocorticoids antagonize AP-1 by inhibiting the Activation/phosphorylation of JNK without affecting its subcellular distribution. J. Cell. Biol. 2000, 150, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Samoilov, M.O.; Rybnikova, E.A.; Sitnik, N.A.; Glushchenko, T.S.; Tyulkova, E.I.; Grinkevich, L.N. Preconditioning modifies the activities of mitogen-activated protein kinases and c-Jun transcription factor in rat hippocampus after severe hypobaric hypoxia. Neurochem. J. 2007, 1, 219–226. [Google Scholar] [CrossRef]
- Mishra, M.; Hedna, V.S. Neuroinflammation after acute ischemic stroke: A volcano hard to contain. Chin. J. Contemp. Neurol. Neurosurg. 2013, 13, 964–970. [Google Scholar] [CrossRef]
- Baschant, U.; Tuckermann, J. The role of the glucocorticoid receptor in inflammation and immunity. J. Steroid Biochem. Mol. Biol. 2010, 120, 69–75. [Google Scholar] [CrossRef]
- Elsby, L.M.; Donn, R.; Alourfi, Z.; Green, L.M.; Beaulieu, E.; Ray, D.W. Hypoxia and glucocorticoid signaling converge to regulate macrophage migration inhibitory factor gene expression. Arthritis Rheum. 2009, 60, 2220–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhao, J.J.; Lv, Y.Y.; Ding, P.S.; Liu, R.Y. Hypoxia down-regulates glucocorticoid receptor alpha and attenuates the anti-inflammatory actions of dexamethasone in human alveolar epithelial A549 cells. Life Sci. 2009, 85, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Y.-Y.; Song, X.-L.; Cai, H.-Y.; Chen, J.-C.; Song, L.-N.; Yang, R.; Lu, J. Upregulations of glucocorticoid-induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J. Immunol. 2012, 188, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hue, C.D.; Cho, F.S.; Cao, S.; Dale Bass, C.; Meaney, D.; Morrison, B. Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J. Cereb. Blood Flow Metab. 2015, 35, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J.R.; Rogatsky, I. Minireview: Glucocorticoids in Autoimmunity: Unexpected Targets and Mechanisms. Mol. Endocrinol. 2011, 25, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Ahmad, A.; Crupi, R.; Impellizzeri, D.; Morabito, R.; Esposito, E.; Cuzzocrea, S. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J. Endocrinol. 2013, 217, 291–301. [Google Scholar] [CrossRef]
- Salvador, E.; Shityakov, S.; Förster, C. Glucocorticoids and endothelial cell barrier function. Cell. Tissue Res. 2014, 355, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Murayi, R.; Chittiboina, P. Glucocorticoids in the management of peritumoral brain edema: A review of molecular mechanisms. Childs Nerv. Syst. 2016, 32, 2293–2302. [Google Scholar] [CrossRef]
- Luo, Z.C.; Xiao, L.; Nuyt, A.M. Mechanisms of developmental programming of the metabolic syndrome and related disorders. World J. Diabetes 2010, 1, 89–98. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J.; Zhuravin, I.A. Role of Prenatal Hypoxia in Brain Development, Cognitive Functions, and Neurodegeneration. Front. Neurosci. 2018, 12, 825. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Ma, Q.; Dasgupta, C.; Xu, Z.; Zhang, L. Antenatal Hypoxia and Programming of Glucocorticoid Receptor Expression in the Adult Rat Heart. Front. Physiol. 2019, 10, 323. [Google Scholar] [CrossRef]
- Deniz, B.F.; Confortim, H.D.; Miguel, P.M.; Bronauth, L.; Fernandes, I.R.; Muotri, A.R.; Pereira, L.O. High Gestational Folic Acid Supplementation Prevents Hypoxia-Ischemia-Induced Caspase-3 Augmenting Without Changing Synapsin and H3 Methylation Levels In The Rat Hippocampus. Int. J. Dev. Neurosci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Newby, E.A.; Myers, D.A.; Ducsay, C.A. Fetal endocrine and metabolic adaptations to hypoxia: The role of the hypothalamic-pituitary-adrenal axis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E429–E439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, D.A.; Singleton, K.; Kenkel, C.; Kaushal, K.M.; Ducsay, C.A. Gestational hypoxia modulates expression of corticotropin-releasing hormone and arginine vasopressin in the paraventricular nucleus in the ovine fetus. Physiol. Rep. 2016, 4, e12643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunwoodie, S.L. The role of hypoxia in development of the Mammalian embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trollmann, R.; Gassmann, M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. 2009, 31, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.; Sabeur, K.; Maltepe, E.; Ameri, K.; Bayraktar, O.; Rowitch, D.H. Sonic Hedgehog Agonist Protects Against Complex Neonatal Cerebellar Injury. Cerebellum 2018, 17, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonkowsky, J.L.; Son, J.H. Hypoxia and connectivity in the developing vertebrate nervous system. Dis. Model. Mech. 2018, 11, dmm037127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrovoy, O.; Tyulkova, E.; Stratilov, V.; Baranova, K.; Nimiritsky, P.; Makarevich, P.; Rybnikova, E. Long-Term Effects of Prenatal Severe Hypoxia on Central and Peripheral Components of the Glucocorticoid System in Rats. Dev. Neurosci. 2020, 42, 145–158. [Google Scholar] [CrossRef]
- Howell, K.R.; Pillai, A. Effects of prenatal hypoxia on schizophrenia-related phenotypes in heterozygous reeler mice: A gene × environment interaction study. Eur. Neuropsychopharmacol. 2014, 24, 1324–1336. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, P.J.; Xiong, F.; Li, Y.; Zhou, J.; Zhang, L. Fetal Hypoxia Increases Vulnerability of Hypoxic-Ischemic Brain Injury in Neonatal Rats: Role of Glucocorticoid Receptors. Neurobiol. Dis. 2014, 65, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, F.; Lin, T.; Song, M.; Ma, Q.; Martinez, S.R.; Lv, J.; MataGreenwood, E.; Xiao, D.; Xu, Z.; Zhang, L. Antenatal hypoxia induces epigenetic repression of glucocorticoid receptor and promotes ischemic-sensitive phenotype in the developing heart. J. Mol. Cell. Cardiol. 2016, 91, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.R.; Ma, Q.; Dasgupta, C.; Meng, X.; Zhang, L. MicroRNA-210 suppresses glucocorticoid receptor expression in response to hypoxia in fetal rat cardiomyocytes. Oncotarget 2017, 8, 80249–80264. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection I,.n hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016, 89, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalivaeva, N.N.; Belyaev, N.D.; Lewis, D.I.; Pickles, A.R.; Makova, N.Z.; Bagrova, D.I.; Dubrovskaya, N.M.; Plesneva, S.A.; Zhuravin, I.A.; Turner, A.J. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J. Mol. Neurosci. 2012, 46, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.J.; Li, T.; Li, J.; Tang, Y.; Le, W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe) /PS1(A246E) transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci. Ther. 2014, 20, 209–217. [Google Scholar] [CrossRef]
- Vasilev, D.S.; Dubrovskaya, N.M.; Zhuravin, I.A.; Nalivaeva, N.N. Developmental Profile of Brain Neprilysin Expression Correlates with Olfactory Behaviour of Rats. J. Mol. Neurosci. 2021. [Google Scholar] [CrossRef]
- Kadiyala, V.; Patrick, N.M.; Mathieu, W.; Jaime-Frias, R.; Pookhao, N.; An, L.; Smith, C.L. Class I lysine deacetylases facilitate glucocorticoid-induced transcription. J. Biol. Chem. 2013, 288, 28900–28912. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Zuo, Z. Prenatal hypoxia-induced adaptation and neuroprotection that is inducible nitric oxide synthase-dependent. Neurobiol. Dis. 2005, 20, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Giusti, S.; Fiszer de Plazas, S. Neuroprotection by hypoxic preconditioning involves upregulation of hypoxia-inducible factor-1 in a prenatal model of acute hypoxia. J. Neurosci. Res. 2012, 90, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.A.; Sanches, E.F.; Odorcyk, F.; Duran-Carabali, L.E.; Sizonenko, S.V. Pregnancy as a valuable period for preventing hypoxia-ischemia brain damage. Int. J. Dev. Neurosci. 2018, 70, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, Z.; Chen, G.; Liu, M.; Wang, H. Prenatal glucocorticoids exposure and fetal adrenal developmental programming. Toxicology 2019, 428, 152308. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybnikova, E.; Nalivaeva, N. Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia. Int. J. Mol. Sci. 2021, 22, 7982. https://doi.org/10.3390/ijms22157982
Rybnikova E, Nalivaeva N. Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia. International Journal of Molecular Sciences. 2021; 22(15):7982. https://doi.org/10.3390/ijms22157982
Chicago/Turabian StyleRybnikova, Elena, and Natalia Nalivaeva. 2021. "Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia" International Journal of Molecular Sciences 22, no. 15: 7982. https://doi.org/10.3390/ijms22157982
APA StyleRybnikova, E., & Nalivaeva, N. (2021). Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia. International Journal of Molecular Sciences, 22(15), 7982. https://doi.org/10.3390/ijms22157982