
Insulin Signal Transduction Perturbations in Insulin Resistance


{kind=link}
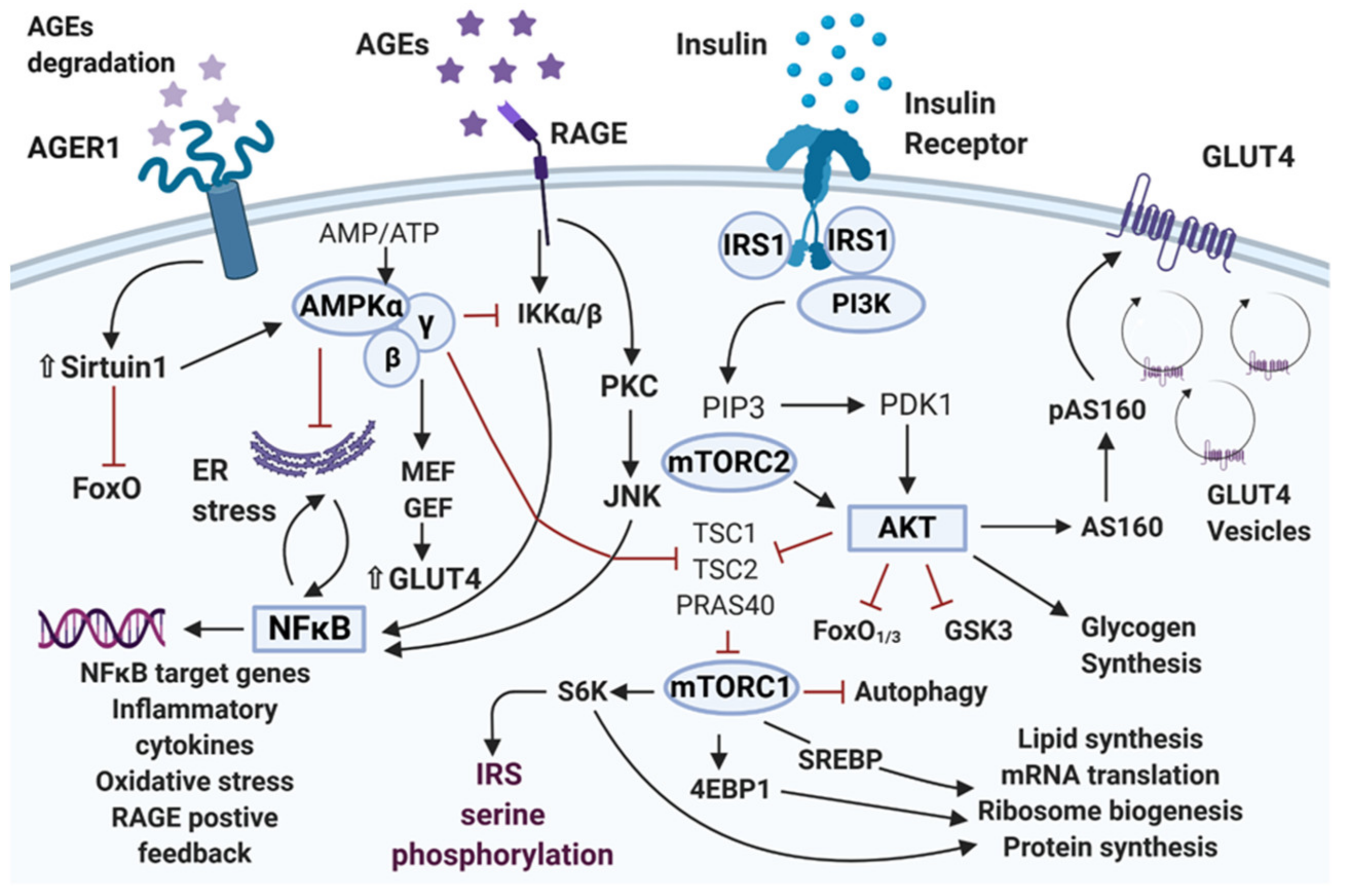
Abstract
:1. Introduction
2. Insulin Receptor and Insulin Signaling Pathway
3. Mechanisms Involved in Insulin Resistance
3.1. β-Cell Function and Mass
3.2. Insulin Receptor Substrate
3.3. Phosphatidylinositol 3-Kinase
3.4. Protein Kinase B/Akt
3.5. GLUT4
3.6. Mammalian Target of Rapamycin/mTOR
3.7. AMP-Activated Protein Kinase
3.8. AGE/RAGE/NF-κB Axis
4. Lipotoxicity, Inflammation, and Oxidative Stress
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnett, D.M.; Krall, L.P. The history of diabetes. In Joslin’s Diabetes Mellitus; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005; pp. 1–17. [Google Scholar]
- Kang, S.; Tsai, L.T.Y.; Rosen, E.D. Nuclear Mechanisms of Insulin Resistance. Trends Cell Biol. 2016, 26, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlinar, B.; Marc, J.; Janež, A.; Pfeifer, M. Molecular Mechanisms of Insulin Resistance and Associated Diseases. Clin. Chim. Acta 2007, 375, 20–35. [Google Scholar] [CrossRef]
- Mezza, T.; Cinti, F.; Cefalo, C.M.A.; Pontecorvi, A.; Kulkarni, R.N.; Giaccari, A. β-Cell Fate in Human Insulin Resistance and Type 2 Diabetes: A Perspective on Islet Plasticity. Diabetes 2019, 68, 1121–1129. [Google Scholar] [CrossRef]
- Muoio, D.M.; Newgard, C.B. Molecular and Metabolic Mechanisms of Insulin Resistance and β-Cell Failure in Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef]
- Fujimoto, W.Y. The Importance of Insulin Resistance in the Pathogenesis of Type 2 Diabetes Mellitus. Am. J. Med. 2000, 108, 9–14. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. AGE Restriction in Diabetes Mellitus: A Paradigm Shift. Nat. Rev. Endocrinol. 2011, 7, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Bril, F.; Cusi, K. Basic concepts in insulin resistance and diabetes treatment. In Dermatology and Diabetes; Springer: Berlin/Heidelberg, Germany, 2018; pp. 19–35. [Google Scholar]
- Khodabandehloo, H.; Gorgani-Firuzjaee, S.; Panahi, G.; Meshkani, R. Molecular and Cellular Mechanisms Linking Inflammation to Insulin Resistance and β-Cell Dysfunction. Transl. Res. 2016, 167, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F. Pathophysiology of diabetes mellitus type 2: Roles of obesity, insulin resistance and β-cell dysfunction. In Diabetes and Cancer; Karger Publishers: Berlin, Germany, 2008; Volume 19, pp. 1–18. [Google Scholar]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-Alpha: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity Plays a Key Role in the Development of Both Insulin Resistance and Muscle Atrophy in Patients with Type 2 Diabetes. Obes. Rev. 2019, 20, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnagarin, R.; Dharmarajan, A.M.; Dass, C.R. Molecular Aspects of Glucose Homeostasis in Skeletal Muscle–A Focus on the Molecular Mechanisms of Insulin Resistance. Mol. Cell. Endocrinol. 2015, 417, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative Stress, Insulin Resistance, Dyslipidemia and Type 2 Diabetes Mellitus. World J. Diabetes 2015, 6, 456. [Google Scholar] [CrossRef]
- Sesti, G.; Federici, M.; Lauro, D.; Sbraccia, P.; Lauro, R. Molecular Mechanism of Insulin Resistance in Type 2 Diabetes Mellitus: Role of the Insulin Receptor Variant Forms. Diabetes/Metab. Res. Rev. 2001, 17, 363–373. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Schinner, S.; Scherbaum, W.A.; Bornstein, S.R.; Barthel, A. Molecular Mechanisms of Insulin Resistance. Diabet. Med. 2005, 22, 674–682. [Google Scholar] [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Rincon, M.; Muzumdar, R.; Atzmon, G.; Barzilai, N. The Paradox of the Insulin/IGF-1 Signaling Pathway in Longevity. Mech. Ageing Dev. 2004, 125, 397–403. [Google Scholar]
- Matheny, R.W., Jr.; Geddis, A.V.; Abdalla, M.N.; Leandry, L.A.; Ford, M.; McClung, H.L.; Pasiakos, S.M. AKT 2 Is the Predominant AKT Isoform Expressed in Human Skeletal Muscle. Physiol. Rep. 2018, 6, e13652. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Prasad, S.K.; Pal, S.; Maji, B.; Syamal, A.K.; Mukherjee, S. Synergistic Protective Effect of Folic Acid and Vitamin B12 against Nicotine-Induced Oxidative Stress and Apoptosis in Pancreatic Islets of the Rat. Pharm. Biol. 2016, 54, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frøsig, C.; Rose, A.J.; Treebak, J.T.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. Effects of Endurance Exercise Training on Insulin Signaling in Human Skeletal Muscle: Interactions at the Level of Phosphatidylinositol 3-Kinase, Akt, and AS160. Diabetes 2007, 56, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Insulin Signaling in Health and Disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [Green Version]
- Cerf, M.E. Beta Cell Dysfunction and Insulin Resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-Cell Deficit and Increased β-Cell Apoptosis in Humans with Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Prigeon, R.L.; McCulloch, D.K.; Boyko, E.J.; Bergman, R.N.; Schwartz, M.W.; Neifing, J.L.; Ward, W.K.; Beard, J.C.; Palmer, J.P. Quantification of the Relationship between Insulin Sensitivity and β-Cell Function in Human Subjects: Evidence for a Hyperbolic Function. Diabetes 1993, 42, 1663–1672. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells during Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Bergman, R.N.; Finegood, D.T.; Kahn, S.E. The Evolution of Β-cell Dysfunction and Insulin Resistance in Type 2 Diabetes. Eur. J. Clin. Investig. 2002, 32, 35–45. [Google Scholar] [CrossRef]
- Kahn, S.E. The Importance of β-Cell Failure in the Development and Progression of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H. Free Fatty Acids Regulate Insulin Secretion from Pancreatic β Cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Mitanchez-Mokhtari, D.; Lahlou, N.; Kieffer, F.; Magny, J.-F.; Roger, M.; Voyer, M. Both Relative Insulin Resistance and Defective Islet β-Cell Processing of Proinsulin Are Responsible for Transient Hyperglycemia in Extremely Preterm Infants. Pediatrics 2004, 113, 537–541. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F. RAGE Binds Preamyloid IAPP Intermediates and Mediates Pancreatic β Cell Proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- LeRoith, D. β-Cell Dysfunction and Insulin Resistance in Type 2 Diabetes: Role of Metabolic and Genetic Abnormalities. Am. J. Med. 2002, 113, 3–11. [Google Scholar] [CrossRef]
- Rabiee, A.; Krüger, M.; Ardenkjær-Larsen, J.; Kahn, C.R.; Emanuelli, B. Distinct Signalling Properties of Insulin Receptor Substrate (IRS)-1 and IRS-2 in Mediating Insulin/IGF-1 Action. Cell. Signal. 2018, 47, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanti, J.-F.; Jager, J. Cellular Mechanisms of Insulin Resistance: Role of Stress-Regulated Serine Kinases and Insulin Receptor Substrates (IRS) Serine Phosphorylation. Curr. Opin. Pharmacol. 2009, 9, 753–762. [Google Scholar] [CrossRef]
- Sharma, M.; Aggarwal, S.; Nayar, U.; Vikram, N.K.; Misra, A.; Luthra, K. Differential Expression of Insulin Receptor Substrate-1 (IRS-1) in Visceral and Subcutaneous Adipose Depots of Morbidly Obese Subjects Undergoing Bariatric Surgery in a Tertiary Care Center in North India; SNP Analysis and Correlation with Metabolic Profile. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 981–986. [Google Scholar]
- Sarah Eckstein, S.; Weigert, C.; Lehmann, R. Divergent Roles of IRS (Insulin Receptor Substrate) 1 and 2 in Liver and Skeletal Muscle. Curr. Med. Chem. 2017, 24, 1827–1852. [Google Scholar] [CrossRef]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.-F. Positive and Negative Regulation of Insulin Signaling through IRS-1 Phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef]
- Suer, F.E.O.; Mergen, H.; Bolu, E.; Ozata, M. Molecular Scanning for Mutations in the Insulin Receptor Substrate-1 (IRS-1) Gene in Turkish with Type 2 Diabetes Mellitus. Endocr. J. 2005, 52, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, I.; Kovacina, K.S.; Roth, R.A. The Insulin Receptor Substrate (IRS)-1 Pleckstrin Homology Domain Functions in Downstream Signaling. J. Biol. Chem. 2001, 276, 8073–8078. [Google Scholar] [CrossRef] [Green Version]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein Kinases: Mechanisms and Downstream Targets in Inflammation-Mediated Obesity and Insulin Resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Zand, H.; Morshedzadeh, N.; Naghashian, F. Signaling Pathways Linking Inflammation to Insulin Resistance. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, S307–S309. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.; Li, S.; Li, W.; Yuan, G.; Pan, Y.; Chen, H. Anti-Diabetic Effects of Inonotus Obliquus Polysaccharides in Streptozotocin-Induced Type 2 Diabetic Mice and Potential Mechanism via PI3K-Akt Signal Pathway. Biomed. Pharmacother. 2017, 95, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Wisessaowapak, C.; Watcharasit, P.; Satayavivad, J. Arsenic Disrupts Neuronal Insulin Signaling through Increasing Free PI3K-P85 and Decreasing PI3K Activity. Toxicol. Lett. 2021, 349, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.K.; Joseph, G.Y.; Ofrecio, J.; Olefsky, J.M. Increased P85/55/50 Expression and Decreased Phosphotidylinositol 3-Kinase Activity in Insulin-Resistant Human Skeletal Muscle. Diabetes 2005, 54, 2351–2359. [Google Scholar] [CrossRef] [Green Version]
- Geering, B.; Cutillas, P.R.; Vanhaesebroeck, B. Regulation of Class IA PI3Ks: Is There a Role for Monomeric PI3K Subunits? Biochem. Soc. Trans. 2007, 35, 199–203. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Hale, P.J.; López-Yunez, A.M.; Chen, J.Y. Genome-Wide Meta-Analysis of Genetic Susceptible Genes for Type 2 Diabetes. BMC Syst. Biol. 2012, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeller, R.; Cantley, L.C. Phosphatidylinositol 3-kinase. Bioessays 1994, 16, 565–576. [Google Scholar] [CrossRef]
- Gao, Y.; Moten, A.; Lin, H.-K. Akt: A New Activation Mechanism. Cell Res. 2014, 24, 785. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Harari, P.; Wheeler, D.; Toulany, M. Targeting AKT/PKB to Improve Treatment Outcomes for Solid Tumors. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2020, 819, 111690. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.W.A.; Elliott, B.T. Akt/PKB Activation and Insulin Signaling: A Novel Insulin Signaling Pathway in the Treatment of Type 2 Diabetes. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One Kinase, Many Modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef]
- Randall, T.A.; Gu, C.; Li, X.; Wang, H.; Shears, S.B. A Two-Way Switch for Inositol Pyrophosphate Signaling: Evolutionary History and Biological Significance of a Unique, Bifunctional Kinase/Phosphatase. Adv. Biol. Regul. 2020, 75, 100674. [Google Scholar] [CrossRef]
- Manning, B.D. Insulin Signaling: Inositol Phosphates Get into the Akt. Cell 2010, 143, 861–863. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A. The Inositol Pyrophosphate Pathway in Health and Diseases. Biol. Rev. 2018, 93, 1203–1227. [Google Scholar] [CrossRef]
- Zhu, Q.; Ghoshal, S.; Rodrigues, A.; Gao, S.; Asterian, A.; Kamenecka, T.M.; Barrow, J.C.; Chakraborty, A. Adipocyte-Specific Deletion of Ip6k1 Reduces Diet-Induced Obesity by Enhancing AMPK-Mediated Thermogenesis. J. Clin. Investig. 2016, 126, 4273–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzarri, M.; Dinicola, S.; Cucina, A. Modulation of Both Insulin Resistance and Cancer Growth by Inositol. Curr. Pharm. Des. 2017, 23, 5200–5210. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT Pathway in Obesity and Type 2 Diabetes. Int. J. Biol. Sci. 2018, 14, 1483. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jessen, N. GLUT4 and UBC9 Protein Expression Is Reduced in Muscle from Type 2 Diabetic Patients with Severe Insulin Resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef]
- Saha, S. Association between the Membrane Transporter Proteins and Type 2 Diabetes Mellitus. Expert Rev. Clin. Pharmacol. 2020, 13, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Jaldin-Fincati, J.R.; Pavarotti, M.; Frendo-Cumbo, S.; Bilan, P.J.; Klip, A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol. Metab. 2017, 28, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty Sweet Years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [Green Version]
- Gannon, N.P.; Conn, C.A.; Vaughan, R.A. Dietary Stimulators of GLUT4 Expression and Translocation in Skeletal Muscle: A Mini-review. Mol. Nutr. Food Res. 2015, 59, 48–64. [Google Scholar] [CrossRef]
- Gaster, M.; Staehr, P.; Beck-Nielsen, H.; Schrøder, H.D.; Handberg, A. GLUT4 Is Reduced in Slow Muscle Fibers of Type 2 Diabetic Patients: Is Insulin Resistance in Type 2 Diabetes a Slow, Type 1 Fiber Disease? Diabetes 2001, 50, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, E.B.; Burcelin, R.; Tsao, T.S.; Stenbit, A.E.; Charron, M.J. The Metabolic Consequences of Altered Glucose Transporter Expression in Transgenic Mice. J. Mol. Med. 1996, 74, 639–652. [Google Scholar] [CrossRef]
- Mora, S.; Yang, C.; Ryder, J.W.; Boeglin, D.; Pessin, J.E. The MEF2A and MEF2D Isoforms Are Differentially Regulated in Muscle and Adipose Tissue during States of Insulin Deficiency. Endocrinology 2001, 142, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.F.; Sparling, D.P.; Olson, A.L.; Winder, W.W.; Dohm, G.L. Regulation of Muscle GLUT4 Enhancer Factor and Myocyte Enhancer Factor 2 by AMP-Activated Protein Kinase. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, E1071–E1076. [Google Scholar] [CrossRef]
- Koh, J.-H.; Hancock, C.R.; Han, D.-H.; Holloszy, J.O.; Nair, K.S.; Dasari, S. AMPK and PPARβ Positive Feedback Loop Regulates Endurance Exercise Training-Mediated GLUT4 Expression in Skeletal Muscle. Am. J. Physiol.-Endocrinol. Metab. 2019, 316, E931–E939. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 Glucose Transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, M.; Wang, J.; Gao, T. Nur77 Increases Glucose Transport in Mouse Skeletal Muscle Cells by Activating P38MAPK Under Lipotoxicity. J. Biomater. Tissue Eng. 2020, 10, 1832–1836. [Google Scholar] [CrossRef]
- Atkinson, B.J.; Griesel, B.A.; King, C.D.; Josey, M.A.; Olson, A.L. Moderate GLUT4 Overexpression Improves Insulin Sensitivity and Fasting Triglyceridemia in High-Fat Diet–Fed Transgenic Mice. Diabetes 2013, 62, 2249–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Herbert, T.P. The Role of Mammalian Target of Rapamycin (MTOR) in the Regulation of Pancreatic β-Cell Mass: Implications in the Development of Type-2 Diabetes. Cell. Mol. Life Sci. 2012, 69, 1289–1304. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR Signaling at a Glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Nie, J.; Van Skike, C.E.; Valentine, J.M.; Orr, M.E. Mammalian Target of Rapamycin at the Crossroad between Alzheimer’s Disease and Diabetes. Diabetes Mellit. 2019, 1128, 185–225. [Google Scholar]
- Yoon, M.-S. MTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J. Absence of S6K1 Protects against Age-and Diet-Induced Obesity While Enhancing Insulin Sensitivity. Nature 2004, 431, 200. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y. Sin1 Phosphorylation Impairs MTORC2 Complex Integrity and Inhibits Downstream Akt Signalling to Suppress Tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; DeStefano, M.A.; Oh, W.J.; Wu, C.; Vega-Cotto, N.M.; Finlan, M.; Liu, D.; Su, B.; Jacinto, E. MTOR Complex 2 Regulates Proper Turnover of Insulin Receptor Substrate-1 via the Ubiquitin Ligase Subunit Fbw8. Mol. Cell 2012, 48, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, Insulin Resistance, and the Metabolic Syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Kola, B.; Korbonits, M. AMPK as a Mediator of Hormonal Signalling. J. Mol. Endocrinol. 2010, 44, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, S.L.; Van Denderen, B.J.W.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-Activated Protein Kinase Regulates GLUT4 Transcription by Phosphorylating Histone Deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Julia Xu, X.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A Long-Standing Partnership? Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Yang, R.; Sheu, M.; Chan, D.; Yang, T.; Tsai, K.; Chiang, C.; Liu, S. Advanced Glycation End-products Induce Skeletal Muscle Atrophy and Dysfunction in Diabetic Mice via a RAGE-mediated, AMPK-down-regulated, Akt Pathway. J. Pathol. 2016, 238, 470–482. [Google Scholar] [CrossRef]
- Xu, X.J.; Gauthier, M.-S.; Hess, D.T.; Apovian, C.M.; Cacicedo, J.M.; Gokce, N.; Farb, M.; Valentine, R.J.; Ruderman, N.B. Insulin Sensitive and Resistant Obesity in Humans: AMPK Activity, Oxidative Stress, and Depot-Specific Changes in Gene Expression in Adipose Tissue. J. Lipid Res. 2012, 53, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Xu, X.J.; Lawson, E.; Deoliveira, R.; Brandon, A.E.; Kraegen, E.W.; Ruderman, N.B. Downregulation of AMPK Accompanies Leucine-and Glucose-Induced Increases in Protein Synthesis and Insulin Resistance in Rat Skeletal Muscle. Diabetes 2010, 59, 2426–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.; Prentki, M. AMP Kinase and Malonyl-CoA: Targets for Therapy of the Metabolic Syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK Activation: A Therapeutic Target for Type 2 Diabetes? DiabetesMetab. Syndr. Obes. Targets Ther. 2014, 7, 241. [Google Scholar]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Unoki, H.; Yamagishi, S. Advanced Glycation End Products and Insulin Resistance. Curr. Pharm. Des. 2008, 14, 987–989. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of Advanced Glycation End Products Improves Insulin Resistance in Human Type 2 Diabetes: Potential Role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Donato, R. RAGE in the Pathophysiology of Skeletal Muscle. J. CachexiaSarcopenia Muscle 2018, 9, 1213–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- C Tobon-Velasco, J.; Cuevas, E.; A Torres-Ramos, M. Receptor for AGEs (RAGE) as Mediator of NF-KB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord.-Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Greene, M.W.; Ruhoff, M.S.; Burrington, C.M.; Garofalo, R.S.; Oreña, S.J. TNFα Activation of PKCδ, Mediated by NFκB and ER Stress, Cross-Talks with the Insulin Signaling Cascade. Cell. Signal. 2010, 22, 274–284. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and Molecular Mechanisms of Endothelial Dysfunction in Diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M. Advanced Glycation End Products-Induced Insulin Resistance Involves Repression of Skeletal Muscle GLUT4 Expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular Mechanisms of Insulin Resistance in Humans and Their Potential Links with Mitochondrial Dysfunction. Diabetes 2006, 55 (Suppl. S2), S9–S15. [Google Scholar] [CrossRef] [Green Version]
- Brons, C.; Grunnet, L.G. Skeletal Muscle Lipotoxicity in Insulin Resistance and Type 2 Diabetes: A Causal Mechanism or an Innocent Bystander. Eur. J. Endocrinol. 2017, 176, R67–R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reali, F.; Morine, M.J.; Kahramanoğulları, O.; Raichur, S.; Schneider, H.-C.; Crowther, D.; Priami, C. Mechanistic Interplay between Ceramide and Insulin Resistance. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, J.; Mensink, M.; Hesselink, M.K.; Ekroos, K.; Schrauwen, P. Long-and Medium-Chain Fatty Acids Induce Insulin Resistance to a Similar Extent in Humans despite Marked Differences in Muscle Fat Accumulation. J. Clin. Endocrinol. Metab. 2012, 97, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides in Metabolism: Key Lipotoxic Players. Annu. Rev. Physiol. 2020, 83, 303–330. [Google Scholar] [CrossRef]
- Bandet, C.L.; Mahfouz, R.; Véret, J.; Sotiropoulos, A.; Poirier, M.; Giussani, P.; Campana, M.; Philippe, E.; Blachnio-Zabielska, A.; Ballaire, R. Ceramide Transporter CERT Is Involved in Muscle Insulin Signaling Defects under Lipotoxic Conditions. Diabetes 2018, 67, 1258–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides in Insulin Resistance and Lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Singh, S.; McGuire, D.K.; Vega, G.L.; Roddy, T.; Reilly, D.F.; Castro-Perez, J.; Kozlitina, J.; Scherer, P.E. Relation of Plasma Ceramides to Visceral Adiposity, Insulin Resistance and the Development of Type 2 Diabetes Mellitus: The Dallas Heart Study. Diabetologia 2018, 61, 2570–2579. [Google Scholar] [CrossRef] [Green Version]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; DeFronzo, R.A.; Kirwan, J.P. Plasma Ceramides Are Elevated in Obese Subjects with Type 2 Diabetes and Correlate with the Severity of Insulin Resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Turpin-Nolan, S.M.; Hammerschmidt, P.; Chen, W.; Jais, A.; Timper, K.; Awazawa, M.; Brodesser, S.; Brüning, J.C. CerS1-Derived C18: 0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep. 2019, 26, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Huang, S.; Wang, X.; Zhang, Q.; Liu, J.; Leng, Y. Downregulation of Lipin-1 Induces Insulin Resistance by Increasing Intracellular Ceramide Accumulation in C2C12 Myotubes. Int. J. Biol. Sci. 2017, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic Ceramides Dissociate Steatosis and Insulin Resistance in Patients with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wågsäter, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Häkkinen, A.-M.; Lindell, M.; Bergholm, R. Adipose Tissue Inflammation and Increased Ceramide Content Characterize Subjects with High Liver Fat Content Independent of Obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, P.; Hames, K.; Leachman, E.; DeLany, J.; Ritov, V.; Menshikova, E.; Dube, J.; Stefanovic-Racic, M.; Toledo, F.; Goodpaster, B. Reduced Skeletal Muscle Oxidative Capacity and Elevated Ceramide but Not Diacylglycerol Content in Severe Obesity. Obesity 2013, 21, 2362–2371. [Google Scholar] [CrossRef] [Green Version]
- Coope, A.; Torsoni, A.S.; Velloso, L.A. Mechanisms in Endocrinology: Metabolic and Inflammatory Pathways on the Pathogenesis of Type 2 Diabetes. Eur. J. Endocrinol. 2016, 174, R175–R187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation, Metaflammation and Immunometabolic Disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid Signaling and Lipotoxicity in Metaflammation: Indications for Metabolic Disease Pathogenesis and Treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, X.; Yang, Y.; He, J.; Gu, H.; Jiang, M.; Huang, Y.; Liu, X.; Liu, L. Resveratrol Inhibits the Development of Obesity-Related Osteoarthritis via the TLR4 and PI3K/Akt Signaling Pathways. Connect. Tissue Res. 2019, 60, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Billiar, T.R.; Machida, K.; Crispe, I.N.; Seki, E. Toll-like Receptor Signaling in Liver Diseases. Gastroenterol. Res. Pract. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benomar, Y.; Taouis, M. Molecular Mechanisms Underlying Obesity-Induced Hypothalamic Inflammation and Insulin Resistance: Pivotal Role of Resistin/TLR4 Pathways. Front. Endocrinol. 2019, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R. Lipid-Induced Insulin Resistance Mediated by the Proinflammatory Receptor TLR4 Requires Saturated Fatty Acid–Induced Ceramide Biosynthesis in Mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A Central Role for JNK in Obesity and Insulin Resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Becattini, B.; Zani, F.; Breasson, L.; Sardi, C.; Giuseppe D’Agostino, V.; Choo, M.; Provenzani, A.; Mo Park, J.; Solinas, G. JNK1 Ablation in Mice Confers Long-term Metabolic Protection from Diet-induced Obesity at the Cost of Moderate Skin Oxidative Damage. FASEB J. 2016, 30, 3124–3132. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Becattini, B. JNK at the Crossroad of Obesity, Insulin Resistance, and Cell Stress Response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β Links Inflammation to Obesity-Induced Insulin Resistance. Nat. Med. 2005, 11, 191. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Endoplasmic Reticulum Stress in Obesity and Diabetes. Int. J. Obes. 2008, 32, S52–S54. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The Molecular Basis for Oxidative Stress-Induced Insulin Resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martinez de Maranon, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I. Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrle, S.; Hsu, W.H. The Etiology of Oxidative Stress in Insulin Resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Zou, X.; Shi, Y.; Liu, Q.; Huyan, T.; Su, J.; Wang, Q.; Zhang, F.; Li, X. FOXO1 Inhibition Prevents Renal Ischemia—Reperfusion Injury via CAMP-response Element Binding Protein/PPAR-γ Coactivator-1α-mediated Mitochondrial Biogenesis. Br. J. Pharmacol. 2020, 177, 432–448. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, M.; Alkaabi, J.; Khan, M.A.B.; Adem, A. Insulin Signal Transduction Perturbations in Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 8590. https://doi.org/10.3390/ijms22168590
Khalid M, Alkaabi J, Khan MAB, Adem A. Insulin Signal Transduction Perturbations in Insulin Resistance. International Journal of Molecular Sciences. 2021; 22(16):8590. https://doi.org/10.3390/ijms22168590
Chicago/Turabian StyleKhalid, Mariyam, Juma Alkaabi, Moien A. B. Khan, and Abdu Adem. 2021. "Insulin Signal Transduction Perturbations in Insulin Resistance" International Journal of Molecular Sciences 22, no. 16: 8590. https://doi.org/10.3390/ijms22168590
APA StyleKhalid, M., Alkaabi, J., Khan, M. A. B., & Adem, A. (2021). Insulin Signal Transduction Perturbations in Insulin Resistance. International Journal of Molecular Sciences, 22(16), 8590. https://doi.org/10.3390/ijms22168590