
GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease

 , , ,
, , ,
Abstract
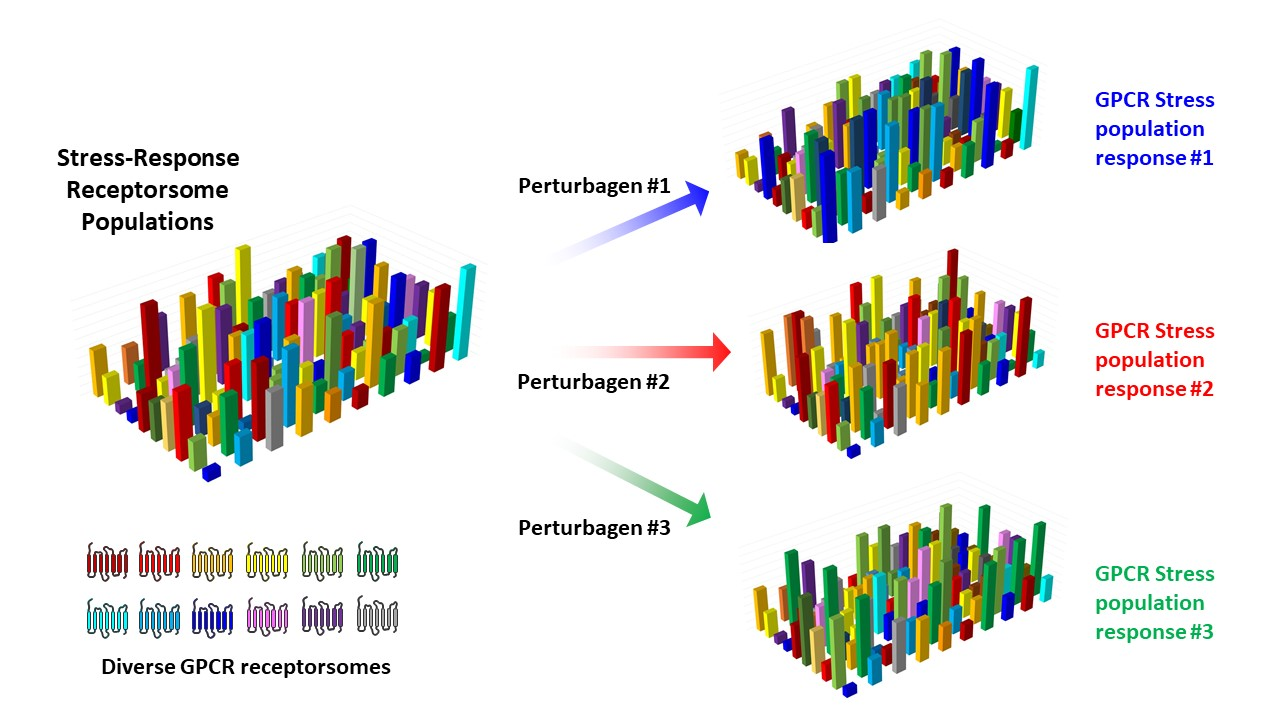
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. G Protein-Coupled Receptors
1.2. Signaling Diversity in GPCRs
1.2.1. G Protein and Non-G Protein Signaling
1.2.2. G Protein-Coupled Receptor Complexes
1.2.3. G Protein Signaling, Endocytosis and Cellular Location
2. Complex Biological Systems
2.1. Functional Properties of Complex Systems
2.1.1. Networks in Pharmacological Systems
2.1.2. Modulation of Networks in Disease and Aging
2.1.3. The Receptors Dilemma and Network Functionality
2.2. Intersection of Systemic GPCR Pharmacology with Complex Systems
2.3. G Protein-Coupled Receptors as System-Level Regulators
3. Pharmacological Interventions within Complex Disease Systems
3.1. Homeostasis and Allostasis within Networks—The Role of GPCRs
3.2. Disease Signatures at the Subcellular Level
3.3. Precision GPCR Interventions for Complex Systems
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- van Gastel, J.; Leysen, H.; Boddaert, J.; Vangenechten, L.; Luttrell, L.M.; Martin, B.; Maudsley, S. Aging-related modifications to G protein-coupled receptor signaling diversity. Pharmacol. Ther. 2021, 223, 107793. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.; Etienne, H.; Idriss, S.; Azmi, A.; Martin, B.; Maudsley, S. Systems-Level G Protein-Coupled Receptor Therapy Across a Neurodegenerative Continuum by the GLP-1 Receptor System. Front. Endocrinol. 2014, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.H.; Wong, Y.H. GPCRs in Autocrine and Paracrine Regulations. Front. Endocrinol. 2019, 10, 428. [Google Scholar] [CrossRef]
- de Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.A.; Vieira, S.I. G(i/o)-Protein Coupled Receptors in the Aging Brain. Front. Aging Neurosci. 2019, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Castillo, J.; Katz, B. Interaction at end-plate receptors between different choline derivatives. Proc. R. Soc. London. Ser. B Biol. Sci. 1957, 146, 369–381. [Google Scholar] [CrossRef]
- Stephenson, R.P. A modification of receptor theory. Br. J. Pharmacol. Chemother. 1956, 11, 379–393. [Google Scholar] [CrossRef]
- De Lean, A.; Stadel, J.M.; Lefkowitz, R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980, 255, 7108–7117. [Google Scholar] [CrossRef]
- Gardella, T.J.; Luck, M.D.; Jensen, G.S.; Schipani, E.; Potts, J.T., Jr.; Jüppner, H. Inverse agonism of amino-terminally truncated parathyroid hormone (PTH) and PTH-related peptide (PTHrP) analogs revealed with constitutively active mutant PTH/PTHrP receptors. Endocrinology 1996, 137, 3936–3941. [Google Scholar] [CrossRef] [Green Version]
- Gether, U.; Lin, S.; Kobilka, B.K. Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J. Biol. Chem. 1995, 270, 28268–28275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leff, P. The two-state model of receptor activation. Trends Pharmacol. Sci. 1995, 16, 89–97. [Google Scholar] [CrossRef]
- Parma, J.; Duprez, L.; Van Sande, J.; Cochaux, P.; Gervy, C.; Mockel, J.; Dumont, J.; Vassart, G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993, 365, 649–651. [Google Scholar] [CrossRef]
- Pozvek, G.; Hilton, J.M.; Quiza, M.; Houssami, S.; Sexton, P.M. Structure/function relationships of calcitonin analogues as agonists, antagonists, or inverse agonists in a constitutively activated receptor cell system. Mol. Pharmacol. 1997, 51, 658–665. [Google Scholar] [CrossRef]
- Shenker, A.; Laue, L.; Kosugi, S.; Merendino, J.J., Jr.; Minegishi, T.; Cutler, G.B., Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 1993, 365, 652–654. [Google Scholar] [CrossRef] [Green Version]
- Samama, P.; Cotecchia, S.; Costa, T.; Lefkowitz, R.J. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993, 268, 4625–4636. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Limbird, L.E. Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of alpha 2-adrenergic receptor-G-protein interactions. A negative charge at amino acid residue 79 forecasts alpha 2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. J. Biol. Chem. 1994, 269, 29557–29564. [Google Scholar] [PubMed]
- Morin, D.; Cotte, N.; Balestre, M.N.; Mouillac, B.; Manning, M.; Breton, C.; Barberis, C. The D136A mutation of the V2 vasopressin receptor induces a constitutive activity which permits discrimination between antagonists with partial agonist and inverse agonist activities. FEBS Lett. 1998, 441, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Alewijnse, A.E.; Timmerman, H.; Jacobs, E.H.; Smit, M.J.; Roovers, E.; Cotecchia, S.; Leurs, R. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol. Pharmacol. 2000, 57, 890–898. [Google Scholar]
- Pauwels, P.J.; Tardif, S.; Wurch, T.; Colpaert, F.C. Facilitation of constitutive alpha(2A)-adrenoceptor activity by both single amino acid mutation (Thr(373)Lys) and g(alphao) protein coexpression: Evidence for inverse agonism. J. Pharmacol. Exp. Ther. 2000, 292, 654–663. [Google Scholar] [PubMed]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Chan, R.; de Maturana, R.L.; Millar, R.P. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004, 64, 7533–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, B.R.; Bourne, H.R. Structural elements of G alpha subunits that interact with G beta gamma, receptors, and effectors. Cell 1993, 73, 631–641. [Google Scholar] [CrossRef]
- Ernst, O.P.; Hofmann, K.P.; Sakmar, T.P. Characterization of rhodopsin mutants that bind transducin but fail to induce GTP nucleotide uptake. Classification of mutant pigments by fluorescence, nucleotide release, and flash-induced light-scattering assays. J. Biol. Chem. 1995, 270, 10580–10586. [Google Scholar] [CrossRef] [Green Version]
- Farahbakhsh, Z.T.; Hideg, K.; Hubbell, W.L. Photoactivated conformational changes in rhodopsin: A time-resolved spin label study. Science 1993, 262, 1416–1419. [Google Scholar] [CrossRef]
- Franke, R.R.; Sakmar, T.P.; Graham, R.M.; Khorana, H.G. Structure and function in rhodopsin. Studies of the interaction between the rhodopsin cytoplasmic domain and transducin. J. Biol. Chem. 1992, 267, 14767–14774. [Google Scholar] [CrossRef]
- Donnelly, D.; Maudsley, S.; Gent, J.P.; Moser, R.N.; Hurrell, C.R.; Findlay, J.B. Conserved polar residues in the transmembrane domain of the human tachykinin NK2 receptor: Functional roles and structural implications. Biochem. J. 1999, 339 Pt 1, 55–61. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Santos-Otte, P.; Luttrell, L.M.; Martin, B.; Maudsley, S. β-Arrestin Based Receptor Signaling Paradigms: Potential Therapeutic Targets for Complex Age-Related Disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Wess, J. G-protein-coupled receptors: Molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J. 1997, 11, 346–354. [Google Scholar] [CrossRef]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M.; et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T. Biased Receptor Signaling in Drug Discovery. Pharmacol. Rev. 2019, 71, 267–315. [Google Scholar] [CrossRef] [Green Version]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Struthers, R.S. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [Green Version]
- Sagan, S.; Chassaing, G.; Pradier, L.; Lavielle, S. Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J. Pharmacol. Exp. Ther. 1996, 276, 1039–1048. [Google Scholar]
- Maudsley, S.; Leysen, H.; van Gastel, J.; Martin, B. Systems Pharmacology: Enabling Multidimensional Therapeutics; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Coffa, S.; Breitman, M.; Spiller, B.W.; Gurevich, V.V. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 2011, 50, 6951–6958. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. Arrestins and G proteins in cellular signaling: The coin has two sides. Sci. Signal. 2018, 11, 549. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef]
- Maudsley, S.; Martin, B.; Luttrell, L.M. The origins of diversity and specificity in g protein-coupled receptor signaling. J. Pharmacol. Exp. Ther. 2005, 314, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudsley, S.; Martin, B.; Gesty-Palmer, D.; Cheung, H.; Johnson, C.; Patel, S.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; Lehrmann, E.; et al. Delineation of a conserved arrestin-biased signaling repertoire in vivo. Mol. Pharmacol. 2015, 87, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesty-Palmer, D.; Yuan, L.; Martin, B.; Wood, W.H., 3rd; Lee, M.H.; Janech, M.G.; Tsoi, L.C.; Zheng, W.J.; Luttrell, L.M.; Maudsley, S. β-arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol. Endocrinol. 2013, 27, 296–314. [Google Scholar] [CrossRef] [Green Version]
- Pierce, K.L.; Maudsley, S.; Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 1489–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudsley, S.; Siddiqui, S.; Martin, B. Systems analysis of arrestin pathway functions. Prog. Mol. Biol. Transl. Sci. 2013, 118, 431–467. [Google Scholar] [CrossRef]
- Pandey, S.; Kumari, P.; Baidya, M.; Kise, R.; Cao, Y.; Dwivedi-Agnihotri, H.; Banerjee, R.; Li, X.X.; Cui, C.S.; Lee, J.D.; et al. Intrinsic bias at non-canonical, β-arrestin-coupled seven transmembrane receptors. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Bockaert, J.; Dumuis, A.; Fagni, L.; Marin, P. GPCR-GIP networks: A first step in the discovery of new therapeutic drugs? Curr. Opin. Drug Discov. Dev. 2004, 7, 649–657. [Google Scholar]
- Eo, H.S.; Choi, J.P.; Noh, S.J.; Hur, C.G.; Kim, W. A combined approach for the classification of G protein-coupled receptors and its application to detect GPCR splice variants. Comput. Biol. Chem. 2007, 31, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C.; von Zastrow, M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 537–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreienkamp, H.J. Organisation of G-protein-coupled receptor signalling complexes by scaffolding proteins. Curr. Opin. Pharmacol. 2002, 2, 581–586. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Maudsley, S.; Patel, S.A.; Park, S.S.; Luttrell, L.M.; Martin, B. Functional signaling biases in G protein-coupled receptors: Game Theory and receptor dynamics. Mini Rev. Med. Chem. 2012, 12, 831–840. [Google Scholar] [CrossRef]
- Marti-Solano, M.; Crilly, S.E.; Malinverni, D.; Munk, C.; Harris, M.; Pearce, A.; Quon, T.; Mackenzie, A.E.; Wang, X.; Peng, J.; et al. Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature 2020, 587, 650–656. [Google Scholar] [CrossRef]
- Maudsley, S.; Martin, B.; Janssens, J.; Etienne, H.; Jushaj, A.; van Gastel, J.; Willemsen, A.; Chen, H.; Gesty-Palmer, D.; Luttrell, L.M. Informatic deconvolution of biased GPCR signaling mechanisms from in vivo pharmacological experimentation. Methods 2016, 92, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Stoeber, M.; Jullié, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; von Zastrow, M. A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action. Neuron 2018, 98, 963–976.e965. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Harmon, C.M. Molecular signatures of human melanocortin receptors for ligand binding and signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2436–2447. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D. The role of structural dynamics in GPCR-mediated signaling. FEBS J. 2021, 288, 2461–2489. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Downey, W.E., 3rd; Colapietro, A.M.; Barak, L.S.; Ménard, L.; Caron, M.G. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 1996, 271, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Sun, H.; Koch, W.J.; Rau, T.; Eschenhagen, T.; Ravens, U.; Heubach, J.F.; Adamson, D.L.; Harding, S.E. Specific beta(2)AR blocker ICI 118,551 actively decreases contraction through a G(i)-coupled form of the beta(2)AR in myocytes from failing human heart. Circulation 2002, 105, 2497–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, R.; Gether, U.; Wenzel-Seifert, K.; Kobilka, B.K. Effects of guanine, inosine, and xanthine nucleotides on beta(2)-adrenergic receptor/G(s) interactions: Evidence for multiple receptor conformations. Mol. Pharmacol. 1999, 56, 348–358. [Google Scholar] [CrossRef]
- Carneiro, D.G.; Clarke, T.; Davies, C.C.; Bailey, D. Identifying novel protein interactions: Proteomic methods, optimisation approaches and data analysis pipelines. Methods 2016, 95, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Chadwick, W.; Janssens, J.; Premont, R.T.; Schmalzigaug, R.; Becker, K.G.; Lehrmann, E.; Wood, W.H.; Zhang, Y.; Siddiqui, S.; et al. GIT2 Acts as a Systems-Level Coordinator of Neurometabolic Activity and Pathophysiological Aging. Front. Endocrinol. 2016, 6, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Martin, B.; Veenker, L.; Beuning, S.; Coppens, V.; Morrens, M.; Maudsley, S. Enhanced Molecular Appreciation of Psychiatric Disorders through High-Dimensionality Data Acquisition and Analytics. Methods Mol. Biol. 2019, 2011, 671–723. [Google Scholar] [CrossRef]
- Bugge, K.; Brakti, I.; Fernandes, C.B.; Dreier, J.E.; Lundsgaard, J.E.; Olsen, J.G.; Skriver, K.; Kragelund, B.B. Interactions by Disorder—A Matter of Context. Front. Mol. Biosci. 2020, 7, 110. [Google Scholar] [CrossRef]
- Randhawa, V.; Pathania, S. Advancing from protein interactomes and gene co-expression networks towards multi-omics-based composite networks: Approaches for predicting and extracting biological knowledge. Brief. Funct. Genom. 2020, 19, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Kalita, B.; Bano, S.; Vavachan, V.M.; Taunk, K.; Seshadri, V.; Rapole, S. Application of mass spectrometry based proteomics to understand diabetes: A special focus on interactomics. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140469. [Google Scholar] [CrossRef]
- Ahn, S.; Maudsley, S.; Luttrell, L.M.; Lefkowitz, R.J.; Daaka, Y. Src-mediated tyrosine phosphorylation of dynamin is required for beta2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J. Biol. Chem. 1999, 274, 1185–1188. [Google Scholar] [CrossRef] [Green Version]
- Rocca, G.J.D.; Maudsley, S.; Daaka, Y.; Lefkowitz, R.J.; Luttrell, L.M. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J. Biol. Chem. 1999, 274, 13978–13984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M. ‘Location, location, location’: Activation and targeting of MAP kinases by G protein-coupled receptors. J. Mol. Endocrinol. 2003, 30, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Sayers, N.; Hanyaloglu, A.C. Intracellular Follicle-Stimulating Hormone Receptor Trafficking and Signaling. Front. Endocrinol. 2018, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Stäubert, C.; Schöneberg, T. GPCR Signaling From Intracellular Membranes—A Novel Concept. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, J.; Leysen, H.; Santos-Otte, P.; Hendrickx, J.O.; Azmi, A.; Martin, B.; Maudsley, S. The RXFP3 receptor is functionally associated with cellular responses to oxidative stress and DNA damage. Aging 2019, 11, 11268–11313. [Google Scholar] [CrossRef]
- Retamal, J.S.; Ramírez-García, P.D.; Shenoy, P.A.; Poole, D.P.; Veldhuis, N.A. Internalized GPCRs as Potential Therapeutic Targets for the Management of Pain. Front. Mol. Neurosci. 2019, 12, 273. [Google Scholar] [CrossRef]
- Fisher, D.N.; Pruitt, J.N. Insights from the study of complex systems for the ecology and evolution of animal populations. Curr. Zool. 2020, 66, 1–14. [Google Scholar] [CrossRef]
- Lambiotte, R.; Rosvall, M.; Scholtes, I. From networks to optimal higher-order models of complex systems. Nat. Phys. 2019, 15, 313–320. [Google Scholar] [CrossRef]
- Gignoux, J.; Chérel, G.; Davies, I.D.; Flint, S.R.; Lateltin, E. Emergence and complex systems: The contribution of dynamic graph theory. Ecol. Complex. 2017, 31, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, F. Complexity in biology. Exceeding the limits of reductionism and determinism using complexity theory. EMBO Rep. 2008, 9, 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickx, J.O.; van Gastel, J.; Leysen, H.; Martin, B.; Maudsley, S. High-dimensionality Data Analysis of Pharmacological Systems Associated with Complex Diseases. Pharmacol. Rev. 2020, 72, 191–217. [Google Scholar] [CrossRef]
- Maudsley, S.; Devanarayan, V.; Martin, B.; Geerts, H. Intelligent and effective informatic deconvolution of “Big Data” and its future impact on the quantitative nature of neurodegenerative disease therapy. Alzheimers Dement. 2018, 14, 961–975. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Trischler, A.P.; D’Eleuterio, G.M. Synthesis of recurrent neural networks for dynamical system simulation. Neural Netw. 2016, 80, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompson, J.J.; Jain, A.; LeCun, Y.; Bregler, C. Joint training of a convolutional network and a graphical model for human pose estimation. Adv. Neural Inf. Process. Syst. 2014, 27, 1799–1807. [Google Scholar]
- Helmstaedter, M.; Briggman, K.L.; Turaga, S.C.; Jain, V.; Seung, H.S.; Denk, W. Connectomic reconstruction of the inner plexiform layer in the mouse retina. Nature 2013, 500, 168–174. [Google Scholar] [CrossRef]
- Cerqueira, F.R.; Ricardo, A.M.; de Oliveira, A.P.; Graber, A.; Baumgartner, C. MUMAL2: Improving sensitivity in shotgun proteomics using cost sensitive artificial neural networks and a threshold selector algorithm. BMC Bioinform. 2016, 17, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Arthur, W.B. The Economy as an Evolving Complex System II; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Levins, R. Complex systems. In Organization Stability & Process; Routledge: Oxfordshire, UK, 2017; pp. 73–88. [Google Scholar]
- Wolf, Y.I.; Katsnelson, M.I.; Koonin, E.V. Physical foundations of biological complexity. Proc. Natl. Acad. Sci. USA 2018, 115, E8678. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, C.; Fatima, B.; Maroua, B. Multi-Agent Simulation Collision Avoidance of Complex System: Application to Evacuation Crowd Behavior. Int. J. Ambient. Comput. Intell. IJACI 2018, 9, 43–59. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudsley, S.; Martin, B.; Egan, J.M. To be or not to be—Obese. Endocrinology 2011, 152, 3592–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransson, L.; Franzén, S.; Rosengren, V.; Wolbert, P.; Sjöholm, Å.; Ortsäter, H. β-Cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J. Endocrinol. 2013, 219, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Kreuch, D.; Keating, D.J.; Wu, T.; Horowitz, M.; Rayner, C.K.; Young, R.L. Gut Mechanisms Linking Intestinal Sweet Sensing to Glycemic Control. Front. Endocrinol. 2018, 9, 741. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology 2000, 22, 108–124. [Google Scholar] [CrossRef]
- Stumvoll, M. Control of glycaemia: From molecules to men. Minkowski Lecture 2003. Diabetologia 2004, 47, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretter, F.; Gebicke-Haerter, P.J.; Albus, M.; an der Heiden, U.; Schwegler, H. Systems biology and addiction. Pharmacopsychiatry 2009, 42 (Suppl. 1), S11–S31. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Adaptive homeostasis. Mol. Asp. Med. 2016, 49, 1–7. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Matzner, F. Neuroevolution on the edge of chaos. In Proceedings of the Genetic and Evolutionary Computation Conference, Berlin, Germany, 15–19 July 2017; pp. 465–472. [Google Scholar]
- Topol, E.J. Individualized medicine from prewomb to tomb. Cell 2014, 157, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Geerts, H.; Dacks, P.A.; Devanarayan, V.; Haas, M.; Khachaturian, Z.S.; Gordon, M.F.; Maudsley, S.; Romero, K.; Stephenson, D. Big data to smart data in Alzheimer’s disease: The brain health modeling initiative to foster actionable knowledge. Alzheimers Dement. 2016, 12, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tian, Y.; Zhang, Z. Network biology in medicine and beyond. Circ. Cardiovasc. Genet. 2014, 7, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Ideker, T.; Nussinov, R. Network approaches and applications in biology. PLoS Comput. Biol. 2017, 13, e1005771. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Bopp, T.; Dixit, S.; Knapp, D.; Emperumal, C.P.; Vergidis, P.; Rajalingam, K.; Melcher, A.; Kannan, N. A blood transcriptome-based analysis of disease progression, immune regulation, and symptoms in coronavirus-infected patients. Cell Death Discov. 2020, 6, 141. [Google Scholar] [CrossRef]
- Aydemir, M.N.; Aydemir, H.B.; Korkmaz, E.M.; Budak, M.; Cekin, N.; Pinarbasi, E. Computationally predicted SARS-COV-2 encoded microRNAs target NFKB, JAK/STAT and TGFB signaling pathways. Gene Rep. 2021, 22, 101012. [Google Scholar] [CrossRef]
- Chadwick, W.; Maudsley, S. The devil is in the dose: Complexity of receptor systems and responses. In Hormesis; Springer: Berlin/Heidelberg, Germany, 2010; pp. 95–108. [Google Scholar]
- van Gastel, J.; Boddaert, J.; Jushaj, A.; Premont, R.T.; Luttrell, L.M.; Janssens, J.; Martin, B.; Maudsley, S. GIT2-A keystone in ageing and age-related disease. Ageing Res. Rev. 2018, 43, 46–63. [Google Scholar] [CrossRef]
- Chatzidoukaki, O.; Goulielmaki, E.; Schumacher, B.; Garinis, G.A. DNA Damage Response and Metabolic Reprogramming in Health and Disease. Trends Genet. 2020, 36, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, Y.; Wang, T.; Ma, Y.; Gao, P.; Chen, J.; Chen, Y.; Yang, B.; Jiao, L. Drug-coated balloon for vertebral artery origin stenosis: A pilot study. J. NeuroInterv. Surg. 2021, 13, 827–830. [Google Scholar] [CrossRef]
- Yegorov, Y.E.; Poznyak, A.V.; Nikiforov, N.G.; Sobenin, I.A.; Orekhov, A.N. The Link between Chronic Stress and Accelerated Aging. Biomedicines 2020, 8, 198. [Google Scholar] [CrossRef]
- Belikov, A.V. Age-related diseases as vicious cycles. Ageing Res. Rev. 2019, 49, 11–26. [Google Scholar] [CrossRef]
- Murthy, V.L.; Yu, B.; Wang, W.; Zhang, X.; Alkis, T.; Pico, A.R.; Yeri, A.; Bhupathiraju, S.N.; Bressler, J.; Ballantyne, C.M.; et al. Molecular Signature of Multisystem Cardiometabolic Stress and Its Association with Prognosis. JAMA Cardiol. 2020, 5, 1144–1153. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Lacaze, P.; Saffery, R.; Phung, J.; Parker, E.; Shah, R.; Murray, A.; Woods, R.L.; Ryan, J. Blood DNA methylation signatures to detect dementia prior to overt clinical symptoms. Alzheimers Dement. 2020, 12, e12056. [Google Scholar] [CrossRef]
- Lipsitz, L.A.; Goldberger, A.L. Loss of ‘complexity’ and aging. Potential applications of fractals and chaos theory to senescence. JAMA 1992, 267, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, R. Chaodynamic loss of complexity and self-similarity in cancer. Med. Hypotheses 1999, 52, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Sleimen-Malkoun, R.; Temprado, J.J.; Hong, S.L. Aging induced loss of complexity and dedifferentiation: Consequences for coordination dynamics within and between brain, muscular and behavioral levels. Front. Aging Neurosci. 2014, 6, 140. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Gu, H.; Luo, Q. Sample entropy reveals an age-related reduction in the complexity of dynamic brain. Sci. Rep. 2017, 7, 7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.X.; Thomas, C.E.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef]
- Faner, R.; Cruz, T.; López-Giraldo, A.; Agustí, A. Network medicine, multimorbidity and the lung in the elderly. Eur. Respir. J. 2014, 44, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vundavilli, H.; Tripathi, L.P.; Datta, A.; Mizuguchi, K. Network modeling and inference of peroxisome proliferator-activated receptor pathway in high fat diet-linked obesity. J. Theor. Biol. 2021, 519, 110647. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, H.; Wang, J.; Wu, F.X. Control principles for complex biological networks. Brief. Bioinform. 2019, 20, 2253–2266. [Google Scholar] [CrossRef]
- Luu, J.; Palczewski, K. Human aging and disease: Lessons from age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 2866–2872. [Google Scholar] [CrossRef] [Green Version]
- Caengprasath, N.; Hanyaloglu, A.C. Hardwiring wire-less networks: Spatially encoded GPCR signaling in endocrine systems. Curr. Opin Cell Biol. 2019, 57, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Santos-Otte, P.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. Int. J. Mol. Sci. 2018, 19, 2919. [Google Scholar] [CrossRef] [Green Version]
- Whitwell, H.J.; Bacalini, M.G.; Blyuss, O.; Chen, S.; Garagnani, P.; Gordleeva, S.Y.; Jalan, S.; Ivanchenko, M.; Kanakov, O.; Kustikova, V.; et al. The Human Body as a Super Network: Digital Methods to Analyze the Propagation of Aging. Front. Aging Neurosci. 2020, 12, 136. [Google Scholar] [CrossRef]
- Azeloglu, E.U.; Iyengar, R. Signaling networks: Information flow, computation, and decision making. Cold Spring Harb. Perspect. Biol. 2015, 7, a005934. [Google Scholar] [CrossRef] [Green Version]
- Bertalanffy, L.V. The History and Status of General Systems Theory. Acad. Manag. J. 1972, 15, 407–426. [Google Scholar] [CrossRef]
- Kohl, M.M.; Paulsen, O. The roles of GABAB receptors in cortical network activity. Adv. Pharmacol. 2010, 58, 205–229. [Google Scholar] [CrossRef]
- Gadkar, K.; Kirouac, D.; Parrott, N.; Ramanujan, S. Quantitative systems pharmacology: A promising approach for translational pharmacology. Drug Discov. Today Technol. 2016, 21–22, 57–65. [Google Scholar] [CrossRef]
- Nicholson, D.N.; Greene, C.S. Constructing knowledge graphs and their biomedical applications. Comput. Struct. Biotechnol. J. 2020, 18, 1414–1428. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Ernst, P.; Siu, A.; Weikum, G. KnowLife: A versatile approach for constructing a large knowledge graph for biomedical sciences. BMC Bioinform. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Jin, Z.; Guo, H.; Jin, H.; Zhang, X.; Smith, T.; Luo, J. Constructing biomedical domain-specific knowledge graph with minimum supervision. Knowl. Inf. Syst. 2020, 62, 317–336. [Google Scholar] [CrossRef]
- Yadav, S.; Ekbal, A.; Saha, S.; Kumar, A.; Bhattacharyya, P. Feature assisted stacked attentive shortest dependency path based Bi-LSTM model for protein-protein interaction. Knowl. -Based Syst. 2019, 166, 18–29. [Google Scholar] [CrossRef]
- Blatti, C., 3rd; Emad, A.; Berry, M.J.; Gatzke, L.; Epstein, M.; Lanier, D.; Rizal, P.; Ge, J.; Liao, X.; Sobh, O.; et al. Knowledge-guided analysis of “omics” data using the KnowEnG cloud platform. PLoS Biol. 2020, 18, e3000583. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Chen, Q.; Zeng, Y.; Jiang, M.; Zhang, Y. Applications of Machine Learning in Drug Target Discovery. Curr. Drug Metab. 2020, 21, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Junker, B.H.; Schreiber, F. Analysis of Biological Networks; John Wiley & Sons: Hoboken, NJ, USA, 2008; Volume 2. [Google Scholar]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Werner, T. Bioinformatics applications for pathway analysis of microarray data. Curr. Opin. Biotechnol. 2008, 19, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Iyengar, R. Systems pharmacology: Network analysis to identify multiscale mechanisms of drug action. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Yeganeh, P.N.; Richardson, C.; Saule, E.; Loraine, A.; Taghi Mostafavi, M. Revisiting the use of graph centrality models in biological pathway analysis. BioData Min. 2020, 13, 5. [Google Scholar] [CrossRef]
- Chen, H.; Martin, B.; Daimon, C.M.; Maudsley, S. Effective use of latent semantic indexing and computational linguistics in biological and biomedical applications. Front. Physiol. 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Martin, B.; Daimon, C.M.; Siddiqui, S.; Luttrell, L.M.; Maudsley, S. Textrous!: Extracting semantic textual meaning from gene sets. PLoS ONE 2013, 8, e62665. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G.; et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016, 7, 12846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourquin, J.; Duncan, D.; Shi, Z.; Zhang, B. GLAD4U: Deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012, 13 (Suppl. 8), S20. [Google Scholar] [CrossRef] [Green Version]
- Glaab, E.; Baudot, A.; Krasnogor, N.; Schneider, R.; Valencia, A. EnrichNet: Network-based gene set enrichment analysis. Bioinformatics 2012, 28, i451–i457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzscheck, N.; Söhle, J.; Kristof, B.; Grönniger, E.; Gallinat, S.; Wenck, H.; Winnefeld, M.; Falckenhayn, C.; Kaderali, L. Multi-omics network analysis reveals distinct stages in the human aging progression in epidermal tissue. Aging 2020, 12, 12393–12409. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, J.; Cao, K.; Zhang, J.; Wang, J. Centrality-based pathway enrichment: A systematic approach for finding significant pathways dominated by key genes. BMC Syst. Biol. 2012, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrea, C.; Taghavi, Z.; Bokanizad, B.; Hanoudi, S.; Tagett, R.; Donato, M.; Voichiţa, C.; Drăghici, S. Methods and approaches in the topology-based analysis of biological pathways. Front. Physiol. 2013, 4, 278. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Shojaie, A.; Michailidis, G. Network-based pathway enrichment analysis with incomplete network information. Bioinformatics 2016, 32, 3165–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belsky, D.W.; Caspi, A.; Houts, R.; Cohen, H.J.; Corcoran, D.L.; Danese, A.; Harrington, H.; Israel, S.; Levine, M.E.; Schaefer, J.D.; et al. Quantification of biological aging in young adults. Proc. Natl. Acad. Sci. USA 2015, 112, E4104–E4110. [Google Scholar] [CrossRef] [Green Version]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.; Houts, R.; et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife 2020, 9, e54870. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.L. Network medicine—From obesity to the “diseasome”. N. Engl. J. Med. 2007, 357, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Simonis, N.; Li, Q.R.; Charloteaux, B.; Heuze, F.; Klitgord, N.; Tam, S.; Yu, H.; Venkatesan, K.; Mou, D.; et al. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009, 5, 321. [Google Scholar] [CrossRef] [PubMed]
- Zilocchi, M.; Moutaoufik, M.T.; Jessulat, M.; Phanse, S.; Aly, K.A.; Babu, M. Misconnecting the dots: Altered mitochondrial protein-protein interactions and their role in neurodegenerative disorders. Expert Rev. Proteom. 2020, 17, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.; Zhang, X.; Wang, Q.; Wang, D. Identification of prostate cancer hub genes and therapeutic agents using bioinformatics approach. Cancer Biomark. 2017, 20, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiarizadeh, M.R.; Mirzaei, S.; Norouzi, M.; Sheybani, N.; Sadi, M.S.V. Identification of Gene Modules and Hub Genes Involved in Mastitis Development Using a Systems Biology Approach. Front. Genet. 2020, 11, 722. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Oti, M.; Snel, B.; Huynen, M.A.; Brunner, H.G. Predicting disease genes using protein-protein interactions. J. Med. Genet. 2006, 43, 691–698. [Google Scholar] [CrossRef]
- Gustafsson, M.; Nestor, C.E.; Zhang, H.; Barabási, A.L.; Baranzini, S.; Brunak, S.; Chung, K.F.; Federoff, H.J.; Gavin, A.C.; Meehan, R.R.; et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Moon, S.J.; Park, K.S.; Tagkopoulos, I. Network-based modeling of drug effects on disease module in systemic sclerosis. Sci. Rep. 2020, 10, 13393. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antal, M.A.; Böde, C.; Csermely, P. Perturbation waves in proteins and protein networks: Applications of percolation and game theories in signaling and drug design. Curr. Protein Pept. Sci. 2009, 10, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Sarajlić, A.; Janjić, V.; Stojković, N.; Radak, D.; Pržulj, N. Network topology reveals key cardiovascular disease genes. PLoS ONE 2013, 8, e71537. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Pieper, A.A.; Nussinov, R.; Lee, G.; Bekris, L.; Leverenz, J.B.; Cummings, J.; Cheng, F. Harnessing endophenotypes and network medicine for Alzheimer’s drug repurposing. Med. Res. Rev. 2020, 40, 2386–2426. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S.I. Molecular gerontology: From homeodynamics to hormesis. Curr. Pharm. Des. 2014, 20, 3036–3039. [Google Scholar] [CrossRef] [Green Version]
- Bakula, D.; Aliper, A.M.; Mamoshina, P.; Petr, M.A.; Teklu, A.; Baur, J.A.; Campisi, J.; Ewald, C.Y.; Georgievskaya, A.; Gladyshev, V.N.; et al. Aging and drug discovery. Aging 2018, 10, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, X.; Huang, Z.; Zheng, Y.; Quan, N. Sestrin 2 controls the cardiovascular aging process via an integrated network of signaling pathways. Ageing Res. Rev. 2020, 62, 101096. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wu, S.J.; Dai, W.T.; Li, Y.X.; Li, Y.Y. The human disease network in terms of dysfunctional regulatory mechanisms. Biol. Direct 2015, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhao, L.; Liu, X.; Hao, Z.; Zhou, Y.; Yang, C.; Li, H. Differential co-expression analysis of rheumatoid arthritis with microarray data. Mol. Med. Rep. 2014, 10, 2421–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, K.; Ritter, L. Diseasome: An approach to understanding gene-disease interactions. Annu. Rev. Nurs. Res. 2011, 29, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Janjić, V.; Pržulj, N. Biological function through network topology: A survey of the human diseasome. Brief. Funct. Genom. 2012, 11, 522–532. [Google Scholar] [CrossRef]
- Janjić, V.; Pržulj, N. The Core Diseasome. Mol. Biosyst. 2012, 8, 2614–2625. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.; Boren, J.; Uhlen, M.; Proctor, G.; Aarsland, D.; Mardinoglu, A.; Shoaie, S. Systems Biology Approaches to Understand the Host-Microbiome Interactions in Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 716. [Google Scholar] [CrossRef]
- Zhou, X.; Menche, J.; Barabási, A.L.; Sharma, A. Human symptoms-disease network. Nat. Commun. 2014, 5, 4212. [Google Scholar] [CrossRef] [Green Version]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.L. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oettrich, J.M.; Dao, V.T.; Frijhoff, J.; Kleikers, P.; Casas, A.I.; Hobbs, A.J.; Schmidt, H.H. Clinical relevance of cyclic GMP modulators: A translational success story of network pharmacology. Clin. Pharmacol. Ther. 2016, 99, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Al-Harazi, O.; Al Insaif, S.; Al-Ajlan, M.A.; Kaya, N.; Dzimiri, N.; Colak, D. Integrated Genomic and Network-Based Analyses of Complex Diseases and Human Disease Network. J. Genet. Genom. 2016, 43, 349–367. [Google Scholar] [CrossRef]
- Wang, R.; Ross, C.A.; Cai, H.; Cong, W.N.; Daimon, C.M.; Carlson, O.D.; Egan, J.M.; Siddiqui, S.; Maudsley, S.; Martin, B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front. Physiol. 2014, 5, 231. [Google Scholar] [CrossRef] [Green Version]
- Jalili, M.; Salehzadeh-Yazdi, A.; Yaghmaie, M.; Ghavamzadeh, A.; Alimoghaddam, K. Cancerome: A hidden informative subnetwork of the diseasome. Comput. Biol. Med. 2016, 76, 173–177. [Google Scholar] [CrossRef]
- Rabieian, R.; Abedi, M.; Gheisari, Y. Central Nodes in Protein Interaction Networks Drive Critical Functions in Transforming Growth Factor Beta-1 Stimulated Kidney Cells. Cell J. 2017, 18, 514–531. [Google Scholar] [CrossRef]
- Langhauser, F.; Casas, A.I.; Dao, V.T.; Guney, E.; Menche, J.; Geuss, E.; Kleikers, P.W.M.; López, M.G.; Barabási, A.L.; Kleinschnitz, C.; et al. A diseasome cluster-based drug repurposing of soluble guanylate cyclase activators from smooth muscle relaxation to direct neuroprotection. NPJ Syst. Biol. Appl. 2018, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Sekiya, M.; Hara, N.; Miyashita, A.; Kuwano, R.; Ikeuchi, T.; Iijima, K.M.; Nakaya, A. Disruption of a RAC1-centred network is associated with Alzheimer’s disease pathology and causes age-dependent neurodegeneration. Hum. Mol. Genet. 2020, 29, 817–833. [Google Scholar] [CrossRef]
- Urbach, D.; Moore, J.H. Mining the diseasome. BioData Min. 2011, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems and Their Role in Cellular Senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 18911. [Google Scholar] [CrossRef] [Green Version]
- Stapelberg, N.J.C.; Neumann, D.L.; Shum, D.; Headrick, J.P. Health, pre-disease and critical transition to disease in the psycho-immune-neuroendocrine network: Are there distinct states in the progression from health to major depressive disorder? Physiol. Behav. 2019, 198, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kundu, D.; Kennedy, L.; Meadows, V.; Baiocchi, L.; Alpini, G.; Francis, H. The Dynamic Interplay between Mast Cells, Aging/Cellular Senescence, and Liver Disease. Gene Expr. 2020, 20, 77–88. [Google Scholar] [CrossRef]
- Guo, J.; Zheng, H.J.; Zhang, W.; Lou, W.; Xia, C.; Han, X.T.; Huang, W.J.; Zhang, F.; Wang, Y.; Liu, W.J. Accelerated Kidney Aging in Diabetes Mellitus. Oxidative Med. Cell. Longev. 2020, 2020, 1234059. [Google Scholar] [CrossRef]
- Liu, Y.I.; Wise, P.H.; Butte, A.J. The “etiome”: Identification and clustering of human disease etiological factors. BMC Bioinform. 2009, 10 (Suppl. 2), S14. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Agarwal, P. A pathway-based view of human diseases and disease relationships. PLoS ONE 2009, 4, e4346. [Google Scholar] [CrossRef]
- van Driel, M.A.; Bruggeman, J.; Vriend, G.; Brunner, H.G.; Leunissen, J.A. A text-mining analysis of the human phenome. Eur. J. Hum. Genet. 2006, 14, 535–542. [Google Scholar] [CrossRef]
- Lage, K.; Karlberg, E.O.; Størling, Z.M.; Olason, P.I.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tümer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Q.; Jiang, R. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics 2009, 25, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.; Venkatraman, V.; Thomas, C.T.; Wu, J.C.; Van Eyk, J.E.; Lam, M.P.Y. Identifying High-Priority Proteins Across the Human Diseasome Using Semantic Similarity. J. Proteome Res. 2018, 17, 4267–4278. [Google Scholar] [CrossRef]
- Suthram, S.; Dudley, J.T.; Chiang, A.P.; Chen, R.; Hastie, T.J.; Butte, A.J. Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol. 2010, 6, e1000662. [Google Scholar] [CrossRef]
- Han, H.W.; Hahn, S.; Jeong, H.Y.; Jee, J.H.; Nam, M.O.; Kim, H.K.; Lee, D.H.; Lee, S.Y.; Choi, D.K.; Yu, J.H.; et al. LINCS L1000 dataset-based repositioning of CGP-60474 as a highly potent anti-endotoxemic agent. Sci. Rep. 2018, 8, 14969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litichevskiy, L.; Peckner, R.; Abelin, J.G.; Asiedu, J.K.; Creech, A.L.; Davis, J.F.; Davison, D.; Dunning, C.M.; Egertson, J.D.; Egri, S.; et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst. 2018, 6, 424–443.e427. [Google Scholar] [CrossRef] [Green Version]
- Gendaszewska-Darmach, E.; Drzazga, A.; Koziołkiewicz, M. Targeting GPCRs Activated by Fatty Acid-Derived Lipids in Type 2 Diabetes. Trends Mol. Med. 2019, 25, 915–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Ward, R.; An, S.; Guo, X.X.; Li, W.; Xu, T.R. Biased signalling: The instinctive skill of the cell in the selection of appropriate signalling pathways. Biochem. J. 2015, 470, 155–167. [Google Scholar] [CrossRef]
- Chadwick, W.; Martin, B.; Chapter, M.C.; Park, S.S.; Wang, L.; Daimon, C.M.; Brenneman, R.; Maudsley, S. GIT2 acts as a potential keystone protein in functional hypothalamic networks associated with age-related phenotypic changes in rats. PLoS ONE 2012, 7, e36975. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Cai, H.; Park, S.S.; Siddiqui, S.; Premont, R.T.; Schmalzigaug, R.; Paramasivam, M.; Seidman, M.; Bodogai, I.; Biragyn, A.; et al. Nuclear GIT2 is an ATM substrate and promotes DNA repair. Mol. Cell. Biol. 2015, 35, 1081–1096. [Google Scholar] [CrossRef] [Green Version]
- Obdržálek, J. DAG-width—Connectivity measure for directed graphs. Symposium on Discrete Algorithms. In Proceedings of the Symposium on Discrete Algorithms; ACM-SIAM: Miami, FL, USA, 2006; pp. 814–821. [Google Scholar]
- Bang-Jensen, J.; Gutin, G. Theory, Algorithms and Applications. Springer Monographs in Mathematics; Springer: London, UK, 2007; Volume 101. [Google Scholar]
- Gruber, H. Digraph complexity measures and applications in formal language theory. Discret. Math. Theor. Comput. Sci. 2012, 14. [Google Scholar] [CrossRef]
- Ferranti, D.; Krane, D.; Craft, D. The value of prior knowledge in machine learning of complex network systems. Bioinformatics 2017, 33, 3610–3618. [Google Scholar] [CrossRef] [Green Version]
- Kuijpers, T.; Wolters, J.; Kleinjans, J.C.; Jennen, D.G. DynOVis: A web tool to study dynamic perturbations for capturing dose-over-time effects in biological networks. BMC Bioinform. 2019, 20, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippmann, C.; Ultsch, A.; Lötsch, J. Computational functional genomics-based reduction of disease-related gene sets to their key components. Bioinformatics 2019, 35, 2362–2370. [Google Scholar] [CrossRef]
- Eggan, L.C. Transition graphs and the star-height of regular events. Mich. Math. J. 1963, 10, 385–397. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Dehmer, M. Networks for systems biology: Conceptual connection of data and function. IET Syst. Biol. 2011, 5, 185–207. [Google Scholar] [CrossRef]
- Balasubramanian, K. Integration of graph theory and quantum chemistry for structure-activity relationships. SAR QSAR Environ. Res. 1994, 2, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Bonchev, D. The overall Wiener index a new tool for characterization of molecular topology. J. Chem. Inf. Comput. Sci. 2001, 41, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, Y.; Basavanagoud, B.; Jamil, M.K. Characteristics studies of molecular structures in drugs. Saudi Pharm. J. 2017, 25, 580–586. [Google Scholar] [CrossRef]
- Xu, C.; Ge, L.; Zhang, Y.; Dehmer, M.; Gutman, I. Computational prediction of therapeutic peptides based on graph index. J. Biomed. Inform. 2017, 75, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, M.; Emmert-Streib, F.; Shi, Y. Interrelations of graph distance measures based on topological indices. PLoS ONE 2014, 9, e94985. [Google Scholar] [CrossRef] [Green Version]
- Mowshowitz, A. Entropy and the complexity of graphs. I. An index of the relative complexity of a graph. Bull. Math. Biophys. 1968, 30, 175–204. [Google Scholar] [CrossRef]
- Klavžar, S.; Rajapakse, A.; Gutman, I. The Szeged and the Wiener index of graphs. Appl. Math. Lett. 1996, 9, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, S.; Kaufman, J.S.; MacLehose, R.F. Analytic bounds on causal risk differences in directed acyclic graphs involving three observed binary variables. J. Stat. Plan. Inference 2009, 139, 3473–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Naoum, F.; Godin, C. Algorithmic height compression of unordered trees. J. Theor. Biol. 2016, 389, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.; Robertson, N.; Seymour, P.D.; Thomas, R. Directed tree-width. J. Comb. Theory Ser. B 2001, 82, 138–154. [Google Scholar] [CrossRef] [Green Version]
- Bermond, J.-C.; Cosnard, M.; Pérennes, S. Directed acyclic graphs with the unique dipath property. Theor. Comput. Sci. 2013, 504, 5–11. [Google Scholar] [CrossRef]
- Myerson, R. Game Theory: Analysis of Conflict; Harvard University Press: Cambridge, MA, USA; London, UK, 1991. [Google Scholar]
- Smith, J.M.; Price, G.R. The logic of animal conflict. Nature 1973, 246, 15–18. [Google Scholar] [CrossRef]
- Christodoulou, C.; Banfield, G.; Cleanthous, A. Self-control with spiking and non-spiking neural networks playing games. J. Physiol. -Paris 2010, 104, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Evolution and the Theory of Games; Cambridge University Press: Cambridge, UK, 1981. [Google Scholar]
- Martin, B.; Brenneman, R.; Golden, E.; Walent, T.; Becker, K.G.; Prabhu, V.V.; Wood, W., 3rd; Ladenheim, B.; Cadet, J.L.; Maudsley, S. Growth factor signals in neural cells: Coherent patterns of interaction control multiple levels of molecular and phenotypic responses. J. Biol. Chem. 2009, 284, 2493–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmon, K.S.; Gong, K.; Yi, J.; Thomas, A.; Moore, C.M.; Masuho, I.; Timson, D.J.; Martemyanov, K.A.; Li, Q.J. LGR5 receptor promotes cell-cell adhesion in stem cells and colon cancer cells via the IQGAP1-Rac1 pathway. J. Biol. Chem. 2017, 292, 14989–15001. [Google Scholar] [CrossRef] [Green Version]
- Neumüller, R.A.; Knoblich, J.A. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009, 23, 2675–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpe, M.S.; McSwain, L.F.; Kudyba, K.; Ng, C.L.; Nicholson, J.; Brady, M.; Qian, Y.; Choksi, V.; Hudson, A.G.; Parrott, B.B.; et al. G-protein signaling is required for increasing germline stem cell division frequency in response to mating in Drosophila males. Sci. Rep. 2020, 10, 3888. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, W.; Brenneman, R.; Martin, B.; Maudsley, S. Complex and multidimensional lipid raft alterations in a murine model of Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, 2010, 604792. [Google Scholar] [CrossRef] [Green Version]
- van Gastel, J.; Cai, H.; Cong, W.N.; Chadwick, W.; Daimon, C.; Leysen, H.; Hendrickx, J.O.; De Schepper, R.; Vangenechten, L.; Van Turnhout, J.; et al. Multidimensional informatic deconvolution defines gender-specific roles of hypothalamic GIT2 in aging trajectories. Mech. Ageing Dev. 2019, 184, 111150. [Google Scholar] [CrossRef]
- Biane, C.; Delaplace, F.; Klaudel, H. Networks and games for precision medicine. Biosystems 2016, 150, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Farahmand, S.; Goliaei, S.; Ansari-Pour, N.; Razaghi-Moghadam, Z. GTA: A game theoretic approach to identifying cancer subnetwork markers. Mol. Biosyst. 2016, 12, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Liu, J.; Zhang, Y.; Dehmer, M. Identifying anticancer peptides by using a generalized chaos game representation. J. Math. Biol. 2019, 78, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, H.J. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Löchel, H.F.; Eger, D.; Sperlea, T.; Heider, D. Deep learning on chaos game representation for proteins. Bioinformatics 2020, 36, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Lieberman Aiden, E. The expanding scope of DNA sequencing. Nat. Biotechnol. 2012, 30, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Weng, G.; Bhalla, U.S.; Iyengar, R. Complexity in biological signaling systems. Science 1999, 284, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Golden, E.; Keselman, A.; Stone, M.; Mattson, M.P.; Egan, J.M.; Maudsley, S. Therapeutic perspectives for the treatment of Huntington’s disease: Treating the whole body. Histol. Histopathol. 2008, 23, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Chadwick, W.; Cong, W.N.; Pantaleo, N.; Daimon, C.M.; Golden, E.J.; Becker, K.G.; Wood, W.H., 3rd; Carlson, O.D.; Egan, J.M.; et al. Euglycemic agent-mediated hypothalamic transcriptomic manipulation in the N171-82Q model of Huntington disease is related to their physiological efficacy. J. Biol. Chem. 2012, 287, 31766–31782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buljan, M.; Ciuffa, R.; van Drogen, A.; Vichalkovski, A.; Mehnert, M.; Rosenberger, G.; Lee, S.; Varjosalo, M.; Pernas, L.E.; Spegg, V.; et al. Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases. Mol. Cell 2020, 79, 504–520.e509. [Google Scholar] [CrossRef]
- Mehnert, M.; Ciuffa, R.; Frommelt, F.; Uliana, F.; van Drogen, A.; Ruminski, K.; Gstaiger, M.; Aebersold, R. Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat. Commun. 2020, 11, 3563. [Google Scholar] [CrossRef]
- Chung, N.C.; Choi, H.; Wang, D.; Mirza, B.; Pelletier, A.R.; Sigdel, D.; Wang, W.; Ping, P. Identifying temporal molecular signatures underlying cardiovascular diseases: A data science platform. J. Mol. Cell. Cardiol. 2020, 145, 54–58. [Google Scholar] [CrossRef]
- Maes, M.; Nowak, G.; Caso, J.R.; Leza, J.C.; Song, C.; Kubera, M.; Klein, H.; Galecki, P.; Noto, C.; Glaab, E.; et al. Toward Omics-Based, Systems Biomedicine, and Path and Drug Discovery Methodologies for Depression-Inflammation Research. Mol. Neurobiol. 2016, 53, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Reid, S.P.; Clark, N.R.; Wang, Z.; Fernandez, N.F.; Rouillard, A.D.; Readhead, B.; Tritsch, S.R.; Hodos, R.; Hafner, M.; et al. L1000CDS(2): LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2016, 2, 16015. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.A.; Choi, Y.S.; Lee, E.J.; Singh, S.R.; Kim, S.C.; Chang, S. A pharmacogenomic analysis using L1000CDS(2) identifies BX-795 as a potential anticancer drug for primary pancreatic ductal adenocarcinoma cells. Cancer Lett. 2019, 465, 82–93. [Google Scholar] [CrossRef]
- Hess, B. Periodic patterns in biology. Naturwissenschaften 2000, 87, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Alpár, A.; Harkany, T. Novel insights into the spatial and temporal complexity of hypothalamic organization through precision methods allowing nanoscale resolution. J. Intern. Med. 2018, 284, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Rosbash, M. The implications of multiple circadian clock origins. PLoS Biol. 2009, 7, e62. [Google Scholar] [CrossRef]
- Castelo-Szekely, V.; Gatfield, D. Emerging Roles of Translational Control in Circadian Timekeeping. J. Mol. Biol. 2020, 432, 3483–3497. [Google Scholar] [CrossRef] [Green Version]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Scientific Background on the Nobel Prize in Chemistry. Available online: https://www.nobelprize.org/uploads/2018/06/advanced-chemistryprize2012.pdf (accessed on 24 November 2021).
- Böhme, I.; Beck-Sickinger, A.G. Illuminating the life of GPCRs. Cell Commun. Signal. 2009, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Linderman, J.J. Modeling of G-protein-coupled receptor signaling pathways. J. Biol. Chem. 2009, 284, 5427–5431. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Field, G.D.; Rieke, F. Mechanisms regulating variability of the single photon responses of mammalian rod photoreceptors. Neuron 2002, 35, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Valenzuela, R.; Granado, N.; Moratalla, R.; Labandeira-Garcia, J.L. Aging-related dysregulation of dopamine and angiotensin receptor interaction. Neurobiol. Aging 2014, 35, 1726–1738. [Google Scholar] [CrossRef] [Green Version]
- Daimon, C.M.; Chirdon, P.; Maudsley, S.; Martin, B. The role of Thyrotropin Releasing Hormone in aging and neurodegenerative diseases. Am. J. Alzheimers Dis. 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Salinas, A.L.; Avila-Zozaya, M.; Ugalde-Silva, P.; Hernández-Guzmán, D.A.; Missirlis, F.; Boucard, A.A. Latrophilins: A Neuro-Centric View of an Evolutionary Conserved Adhesion G Protein-Coupled Receptor Subfamily. Front. Neurosci. 2019, 13, 700. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Maudsley, S.; Gesty-Palmer, D. Translating in vitro ligand bias into in vivo efficacy. Cell. Signal. 2018, 41, 46–55. [Google Scholar] [CrossRef]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Freestone, S.H.; de Maturana, R.L.; Thomson, A.A.; Millar, R.P. Gonadotropin-releasing hormone functionally antagonizes testosterone activation of the human androgen receptor in prostate cells through focal adhesion complexes involving Hic-5. Neuroendocrinology 2006, 84, 285–300. [Google Scholar] [CrossRef]
- Tran, Q.K.; VerMeer, M.; Burgard, M.A.; Hassan, A.B.; Giles, J. Hetero-oligomeric Complex between the G Protein-coupled Estrogen Receptor 1 and the Plasma Membrane Ca2+-ATPase 4b. J. Biol. Chem. 2015, 290, 13293–13307. [Google Scholar] [CrossRef] [Green Version]
- Veldhuis, N.A.; Poole, D.P.; Grace, M.; McIntyre, P.; Bunnett, N.W. The G protein-coupled receptor-transient receptor potential channel axis: Molecular insights for targeting disorders of sensation and inflammation. Pharmacol. Rev. 2015, 67, 36–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.M.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A.; Lymperopoulos, A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int. J. Mol. Sci. 2018, 19, 3764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Burg, E.H.; Neumann, I.D. Bridging the gap between GPCR activation and behaviour: Oxytocin and prolactin signalling in the hypothalamus. J. Mol. Neurosci. 2011, 43, 200–208. [Google Scholar] [CrossRef] [PubMed]
- West, C.; Hanyaloglu, A.C. Minireview: Spatial Programming of G Protein-Coupled Receptor Activity: Decoding Signaling in Health and Disease. Mol. Endocrinol. 2015, 29, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Carbone, S.E.; Veldhuis, N.A.; Gondin, A.B.; Poole, D.P. G protein-coupled receptor trafficking and signaling: New insights into the enteric nervous system. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G446–G452. [Google Scholar] [CrossRef]
- Millar, R.P.; Lu, Z.L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-releasing hormone receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Price, D.D.; Mayer, D.J. Mechanisms of hyperalgesia and morphine tolerance: A current view of their possible interactions. Pain 1995, 62, 259–274. [Google Scholar] [CrossRef]
- Whistler, J.L. Examining the role of mu opioid receptor endocytosis in the beneficial and side-effects of prolonged opioid use: From a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012, 121, 189–204. [Google Scholar] [CrossRef] [Green Version]
- Assi, R.; Kotecki, N.; Awada, A. Targeting molecular subtypes in solid cancers: Successes and failures. Curr. Opin. Oncol. 2020, 32, 488–493. [Google Scholar] [CrossRef]
- Alqahtani, A.M.; Kumarappan, C.; Kumar, V.; Srinivasan, R.; Krishnaraju, V. Understanding the genetic aspects of resistance to antidepressants treatment. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7784–7795. [Google Scholar] [CrossRef]
- Kitanaka, N.; Kitanaka, J.; Hall, F.S.; Tatsuta, T.; Morita, Y.; Takemura, M.; Wang, X.B.; Uhl, G.R. Alterations in the levels of heterotrimeric G protein subunits induced by psychostimulants, opiates, barbiturates, and ethanol: Implications for drug dependence, tolerance, and withdrawal. Synapse 2008, 62, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Burford, N.T.; Traynor, J.R.; Alt, A. Positive allosteric modulators of the μ-opioid receptor: A novel approach for future pain medications. Br. J. Pharmacol. 2015, 172, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.L.; Patel, R.; Jayanthi, S. Compulsive methamphetamine taking and abstinence in the presence of adverse consequences: Epigenetic and transcriptional consequences in the rat brain. Pharmacol. Biochem. Behav. 2019, 179, 98–108. [Google Scholar] [CrossRef]
- Cadet, J.L.; Bisagno, V. Neuropsychological Consequences of Chronic Drug Use: Relevance to Treatment Approaches. Front. Psychiatry 2015, 6, 189. [Google Scholar] [CrossRef]
- Yang, D.; Wei, G.; Long, F.; Nie, H.; Tian, X.; Qu, L.; Wang, S.; Li, P.; Qiu, Y.; Wang, Y.; et al. Histone methyltransferase Smyd3 is a new regulator for vascular senescence. Aging Cell 2020, 19, e13212. [Google Scholar] [CrossRef]
- Szymczak, S.; Dose, J.; Torres, G.G.; Heinsen, F.A.; Venkatesh, G.; Datlinger, P.; Nygaard, M.; Mengel-From, J.; Flachsbart, F.; Klapper, W.; et al. DNA methylation QTL analysis identifies new regulators of human longevity. Hum. Mol. Genet. 2020, 29, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Sterling, P.; Eyer, J. Handbook of Life Stress, Cognition and Health. Fisher, S., Reason, J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1988. [Google Scholar]
- Grieder, M.; Wang, D.J.J.; Dierks, T.; Wahlund, L.O.; Jann, K. Default Mode Network Complexity and Cognitive Decline in Mild Alzheimer’s Disease. Front. Neurosci. 2018, 12, 770. [Google Scholar] [CrossRef] [Green Version]
- Lunghi, C.; Consorti, G.; Tramontano, M.; Esteves, J.E.; Cerritelli, F. Perspectives on tissue adaptation related to allostatic load: Scoping review and integrative hypothesis with a focus on osteopathic palpation. J. Bodyw. Mov. Ther. 2020, 24, 212–220. [Google Scholar] [CrossRef]
- Leslie, R.D.; Vartak, T. Allostasis and the origins of adult-onset diabetes. Diabetologia 2020, 63, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Jestin, M.; Kapnick, S.M.; Tarasenko, T.N.; Burke, C.T.; Zerfas, P.M.; Diaz, F.; Vernon, H.; Singh, L.N.; Sokol, R.J.; McGuire, P.J. Mitochondrial disease disrupts hepatic allostasis and lowers the threshold for immune-mediated liver toxicity. Mol. Metab. 2020, 37, 100981. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef]
- Schulkin, J. Allostasis, Homeostasis, and the Costs of Physiological Adaptation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Lee, S.W. A Copernican Approach to Brain Advancement: The Paradigm of Allostatic Orchestration. Front. Hum. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Yoris, A.; Legaz, A.; Abrevaya, S.; Alarco, S.; López Peláez, J.; Sánchez, R.; García, A.M.; Ibáñez, A.; Sedeño, L. Multicentric evidence of emotional impairments in hypertensive heart disease. Sci. Rep. 2020, 10, 14131. [Google Scholar] [CrossRef]
- Paciorek, A.; Skora, L. Vagus Nerve Stimulation as a Gateway to Interoception. Front. Psychol. 2020, 11, 1659. [Google Scholar] [CrossRef]
- Burke, N.N.; Finn, D.P.; McGuire, B.E.; Roche, M. Psychological stress in early life as a predisposing factor for the development of chronic pain: Clinical and preclinical evidence and neurobiological mechanisms. J. Neurosci. Res. 2017, 95, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.W.; Midgley, J.E.M.; Hoermann, R. Editorial: “Homeostasis and Allostasis of Thyroid Function”. Front. Endocrinol. 2018, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Mayer, E.A.; Naliboff, B.D.; Chang, L.; Coutinho, S.V.V. Stress and irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G519–G524. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Schulkin, J. Addiction and stress: An allostatic view. Neurosci. Biobehav. Rev. 2018, 106, 245–262. [Google Scholar] [CrossRef]
- Engel, A.K.; Fries, P.; Singer, W. Dynamic predictions: Oscillations and synchrony in top-down processing. Nat. Rev. Neurosci. 2001, 2, 704–716. [Google Scholar] [CrossRef]
- Buzsaki, G. Rhythms of the Brain; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Kleiger, R.E.; Miller, J.P.; Bigger, J.T., Jr.; Moss, A.J. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am. J. Cardiol. 1987, 59, 256–262. [Google Scholar] [CrossRef]
- Dekker, J.M.; Schouten, E.G.; Klootwijk, P.; Pool, J.; Swenne, C.A.; Kromhout, D. Heart rate variability from short electrocardiographic recordings predicts mortality from all causes in middle-aged and elderly men. The Zutphen Study. Am. J. Epidemiol. 1997, 145, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, H.; Perelman, D.; Breschi, A.; Limcaoco, P.; Kellogg, R.; McLaughlin, T.; Snyder, M. Glucotypes reveal new patterns of glucose dysregulation. PLoS Biol. 2018, 16, e2005143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buysse, D.J. Sleep health: Can we define it? Does it matter? Sleep 2014, 37, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintock, M.K.; Dale, W.; Laumann, E.O.; Waite, L. Empirical redefinition of comprehensive health and well-being in the older adults of the United States. Proc. Natl. Acad. Sci. USA 2016, 113, E3071–E3080. [Google Scholar] [CrossRef] [Green Version]
- Kalisch, R.; Müller, M.B.; Tüscher, O. A conceptual framework for the neurobiological study of resilience. Behav. Brain Sci. 2015, 38, e92. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Laukkanen, M.O.; Vinciguerra, M.; Colangelo, T.; Colantuoni, V. A Timeless Link between Circadian Patterns and Disease. Trends Mol. Med. 2016, 22, 68–81. [Google Scholar] [CrossRef]
- Martin, B.; Chen, H.; Daimon, C.M.; Chadwick, W.; Siddiqui, S.; Maudsley, S. Plurigon: Three dimensional visualization and classification of high-dimensionality data. Front. Physiol. 2013, 4, 190. [Google Scholar] [CrossRef] [Green Version]
- De Ridder, W.; Azmi, A.; Clemen, C.S.; Eichinger, L.; Hofmann, A.; Schröder, R.; Johnson, K.; Töpf, A.; Straub, V.; De Jonghe, P.; et al. Multisystem proteinopathy due to a homozygous p.Arg159His VCP mutation: A tale of the unexpected. Neurology 2020, 94, e785–e796. [Google Scholar] [CrossRef] [Green Version]
- Lum, P.Y.; Singh, G.; Lehman, A.; Ishkanov, T.; Vejdemo-Johansson, M.; Alagappan, M.; Carlsson, J.; Carlsson, G. Extracting insights from the shape of complex data using topology. Sci. Rep. 2013, 3, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahmad, H.F.; Peng, W.; Zhu, R.; Ballout, F.; Monzer, A.; Elajami, M.K.; Kobeissy, F.; Abou-Kheir, W.; Mechref, Y. Protein Expression Analysis of an In Vitro Murine Model of Prostate Cancer Progression: Towards Identification of High-Potential Therapeutic Targets. J. Pers. Med. 2020, 10, 83. [Google Scholar] [CrossRef]
- Lupo, G.; Gaetani, S.; Cacci, E.; Biagioni, S.; Negri, R. Molecular Signatures of the Aging Brain: Finding the Links between Genes and Phenotypes. Neurotherapeutics 2019, 16, 543–553. [Google Scholar] [CrossRef]
- de Jong, S.; Newhouse, S.J.; Patel, H.; Lee, S.; Dempster, D.; Curtis, C.; Paya-Cano, J.; Murphy, D.; Wilson, C.E.; Horder, J.; et al. Immune signatures and disorder-specific patterns in a cross-disorder gene expression analysis. Br. J. Psychiatry 2016, 209, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Lustig, A.; Carter, A.; Sankar, M.; Daimon, C.M.; Premont, R.T.; Etienne, H.; van Gastel, J.; Azmi, A.; Janssens, J.; et al. Genomic deletion of GIT2 induces a premature age-related thymic dysfunction and systemic immune system disruption. Aging 2017, 9, 706–740. [Google Scholar] [CrossRef] [Green Version]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Walsh, J.R.; Long, J.; Davis, C.B.; Henstock, P.; Hodge, M.R.; Maciejewski, M.; Mu, X.J.; Ra, S.; Zhao, S. Standard machine learning approaches outperform deep representation learning on phenotype prediction from transcriptomics data. BMC Bioinform. 2020, 21, 119. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.G.; Nilsson, E.J.; Rutardóttir, S.; Paczesny, J.; Pallon, J.; Akerström, B. Bystander cell death and stress response is inhibited by the radical scavenger α(1)-microglobulin in irradiated cell cultures. Radiat. Res. 2010, 174, 590–600. [Google Scholar] [CrossRef]
- Ishii, M.; Rohrer, B. Bystander effects elicited by single-cell photo-oxidative blue-light stimulation in retinal pigment epithelium cell networks. Cell Death Discov. 2017, 3, 16071. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; The National Academic Press: Washington, DC, USA, 2011. [Google Scholar]
- Sankar, P.L.; Parker, L.S. The Precision Medicine Initiative’s All of Us Research Program: An agenda for research on its ethical, legal, and social issues. Genet. Med. 2017, 19, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegaert, K.; Smits, A.; van Donge, T.; van den Anker, J.; Sarafidis, K.; Levtchenko, E.; Mekahli, D. Renal Precision Medicine in Neonates and Acute Kidney Injury: How to Convert a Cloud of Creatinine Observations to Support Clinical Decisions. Front. Pediatrics 2020, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Sullivan, D.K.; Wells, A.C.; Chen, J.H. ClinicNet: Machine learning for personalized clinical order set recommendations. JAMIA Open 2020, 3, 216–224. [Google Scholar] [CrossRef]
- Bilkey, G.A.; Burns, B.L.; Coles, E.P.; Mahede, T.; Baynam, G.; Nowak, K.J. Optimizing Precision Medicine for Public Health. Front. Public Health 2019, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Saponaro, C.; Sergio, S.; Coluccia, A.; De Luca, M.; La Regina, G.; Mologni, L.; Famiglini, V.; Naccarato, V.; Bonetti, D.; Gautier, C.; et al. β-catenin knockdown promotes NHERF1-mediated survival of colorectal cancer cells: Implications for a double-targeted therapy. Oncogene 2018, 37, 3301–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilter, M.; Mansoor, S.; Sensoy, O. Utilization of Biased G Protein-Coupled ReceptorSignaling towards Development of Safer andPersonalized Therapeutics. Molecules 2019, 24, 2052. [Google Scholar] [CrossRef] [Green Version]
- Nairismägi, M.L.; Tan, J.; Lim, J.Q.; Nagarajan, S.; Ng, C.C.; Rajasegaran, V.; Huang, D.; Lim, W.K.; Laurensia, Y.; Wijaya, G.C.; et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia 2016, 30, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Nisar, S.P.; Jones, M.L.; Cunningham, M.R.; Mumford, A.D.; Mundell, S.J. Rare platelet GPCR variants: What can we learn? Br. J. Pharmacol. 2015, 172, 3242–3253. [Google Scholar] [CrossRef] [Green Version]
- Fukami, M.; Suzuki, E.; Igarashi, M.; Miyado, M.; Ogata, T. Gain-of-function mutations in G-protein-coupled receptor genes associated with human endocrine disorders. Clin. Endocrinol. 2018, 88, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wu, J.; Jia, H.; Wang, X.; Zheng, R.; Jiang, F.; Chen, D.N.; Chen, Z.; Li, J.D. PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: Selective disruption of the binding to a Gα-protein leads to biased signaling. FASEB J. 2019, 33, 4538–4546. [Google Scholar] [CrossRef]
- Kleinau, G.; Müller, A.; Biebermann, H. Oligomerization of GPCRs involved in endocrine regulation. J. Mol. Endocrinol. 2016, 57, R59–R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penlioglou, T.; Stoian, A.P.; Papanas, N. Diabetes, Vascular Aging and Stroke: Old Dogs, New Tricks? J. Clin. Med. 2021, 10, 4620. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Buckley, J.; Cicek, M.; Gregg, E.W. The Changing Nature of Mortality and Morbidity in Patients with Diabetes. Endocrinol. Metab. Clin. N. Am. 2021, 50, 357–368. [Google Scholar] [CrossRef]
- Wagner, K.H.; Schwingshackl, L.; Draxler, A.; Franzke, B. Impact of dietary and lifestyle interventions in elderly or people diagnosed with diabetes, metabolic disorders, cardiovascular disease, cancer and micronutrient deficiency on micronuclei frequency—A systematic review and meta-analysis. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108367. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. 2021, 22, 196–231. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leysen, H.; Walter, D.; Christiaenssen, B.; Vandoren, R.; Harputluoğlu, İ.; Van Loon, N.; Maudsley, S. GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease. Int. J. Mol. Sci. 2021, 22, 13387. https://doi.org/10.3390/ijms222413387
Leysen H, Walter D, Christiaenssen B, Vandoren R, Harputluoğlu İ, Van Loon N, Maudsley S. GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease. International Journal of Molecular Sciences. 2021; 22(24):13387. https://doi.org/10.3390/ijms222413387
Chicago/Turabian StyleLeysen, Hanne, Deborah Walter, Bregje Christiaenssen, Romi Vandoren, İrem Harputluoğlu, Nore Van Loon, and Stuart Maudsley. 2021. "GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease" International Journal of Molecular Sciences 22, no. 24: 13387. https://doi.org/10.3390/ijms222413387
APA StyleLeysen, H., Walter, D., Christiaenssen, B., Vandoren, R., Harputluoğlu, İ., Van Loon, N., & Maudsley, S. (2021). GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease. International Journal of Molecular Sciences, 22(24), 13387. https://doi.org/10.3390/ijms222413387