Conformational Landscape of Cytochrome P450 Reductase Interactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
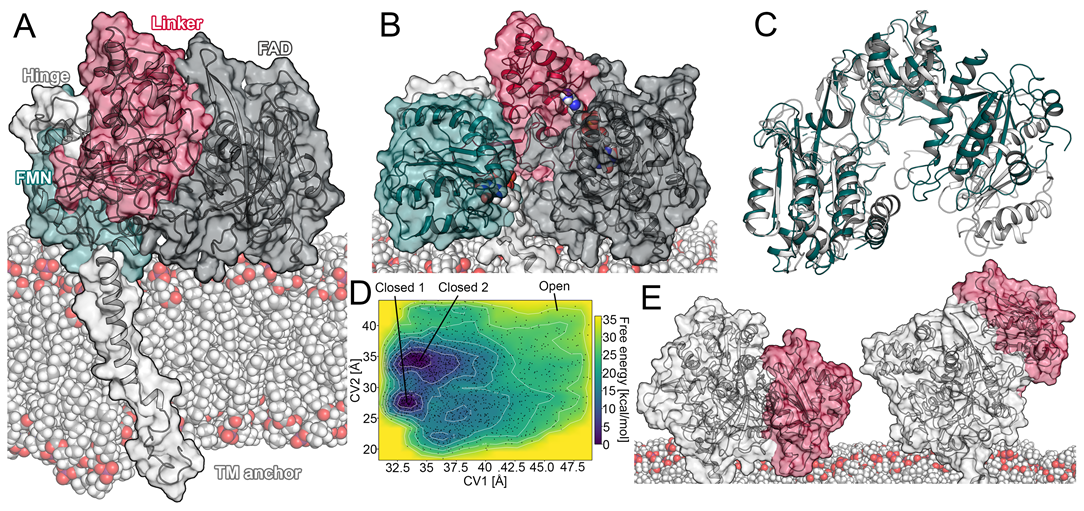
2.1. Open Conformation of the CPR
2.1.1. Transitions between a Sitting and Upright Position
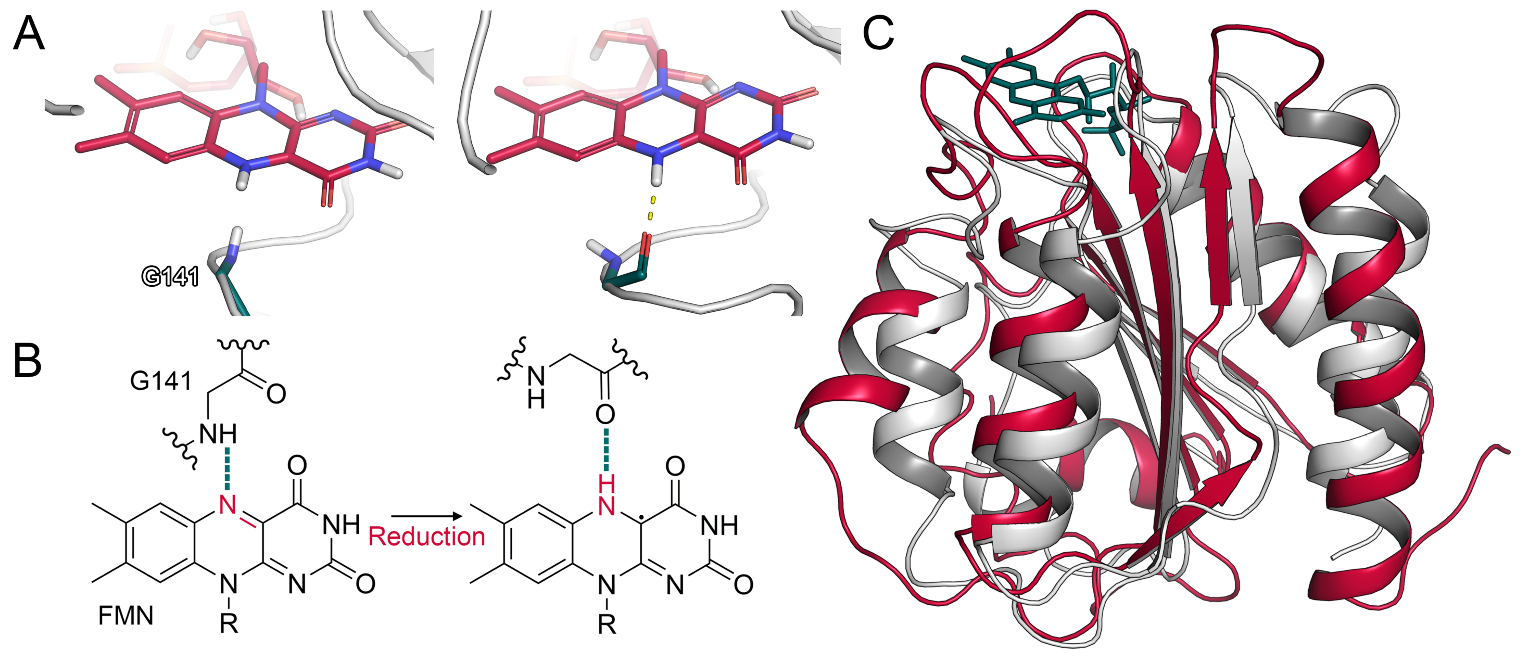
2.1.2. Glycine Flip upon Reduction of FMN
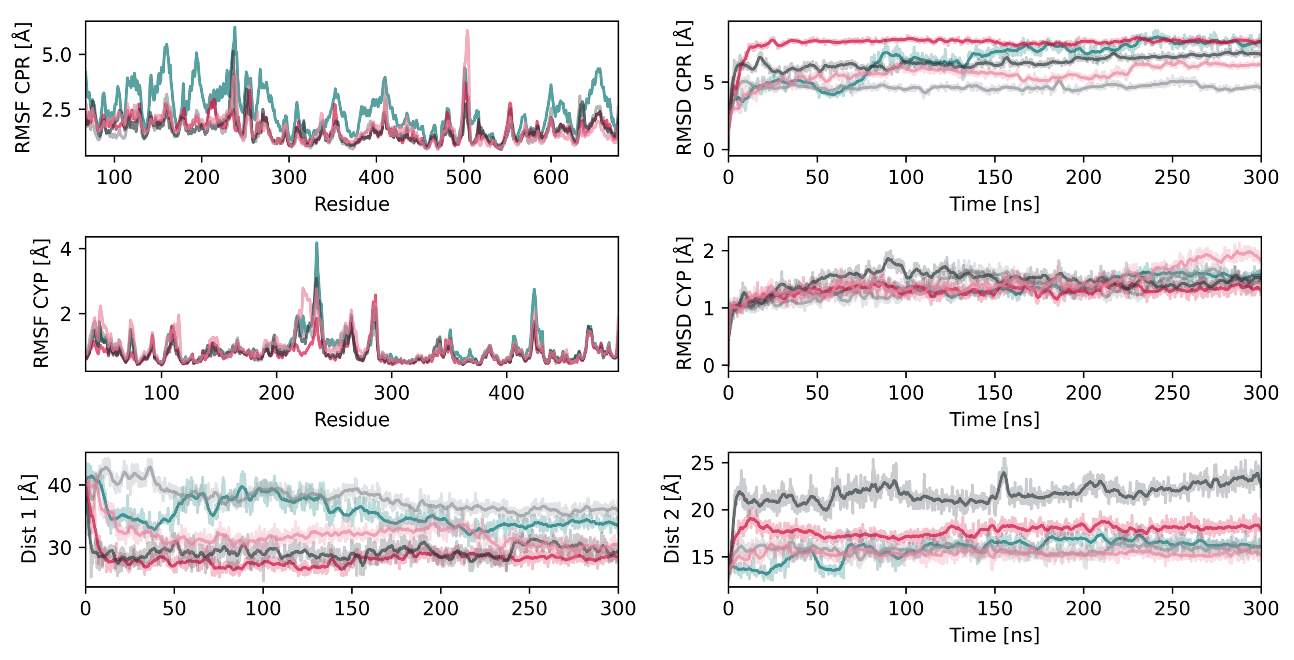
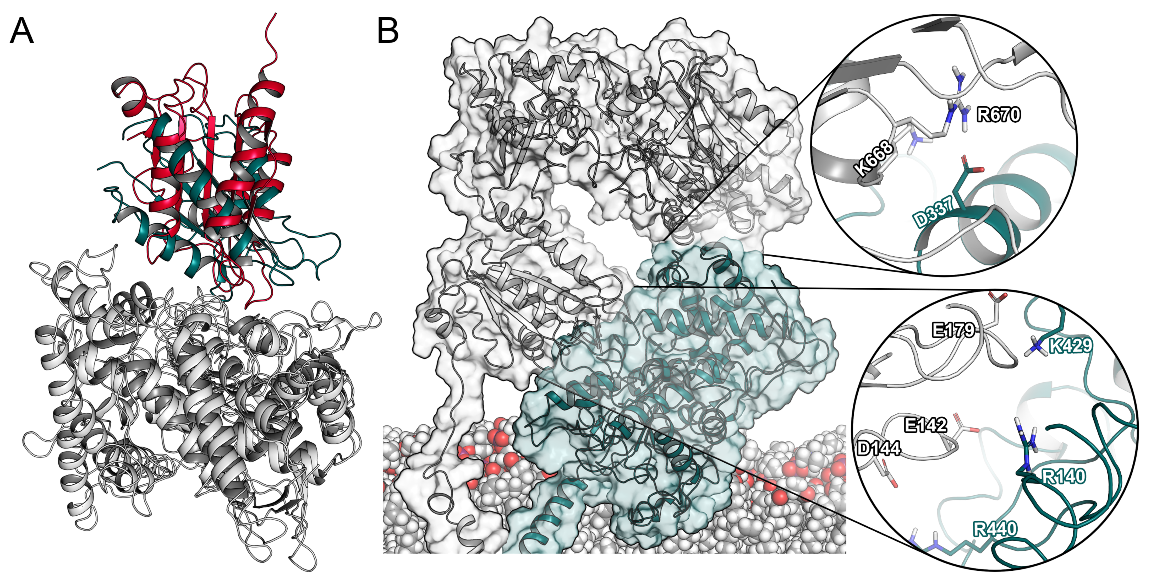
2.2. Interactions between CYP2D6 and the CPR
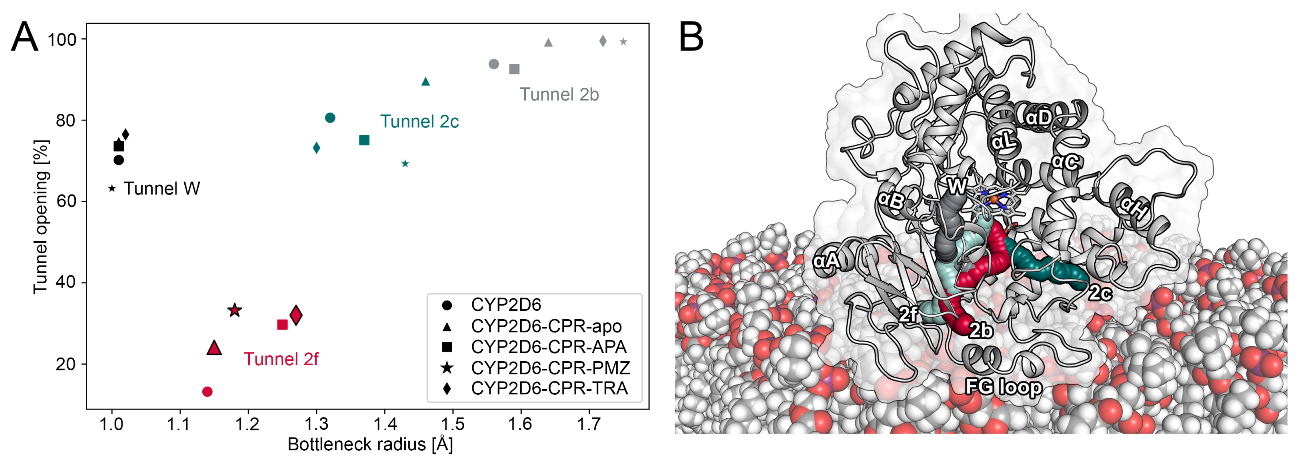
2.3. CPR Binding Affects Tunnels in CYP2D6
3. Materials and Methods
3.1. Simulation Parameters
3.2. Model Building
3.3. Conventional MD Simulations and Post-Processing
3.4. Metadynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furge, L.L.; Guengerich, F.P. Cytochrome P450 enzymes in drug metabolism and chemical toxicology: An introduction. Biochem. Mol. Biol. Educ. 2006, 34, 66–74. [Google Scholar] [CrossRef]
- Schiffer, L.; Barnard, L.; Baranowski, E.S.; Gilligan, L.C.; Taylor, A.E.; Arlt, W.; Shackleton, C.H.; Storbeck, K.H. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2019, 194, 105439. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M. Spontaneous Ligand Access Events to Membrane-Bound Cytochrome P450 2D6 Sampled at Atomic Resolution. Sci. Rep. 2019, 9, 16411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Polymorphism of Human Cytochrome P450 2D6 and Its Clinical Significance. Clin. Pharmacokinet. 2012, 48, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Dubey, K.D.; Shaik, S. Choreography of the Reductase and P450BM3 Domains Toward Electron Transfer Is Instigated by the Substrate. J. Am. Chem. Soc. 2018, 140, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Sündermann, A.; Oostenbrink, C. Molecular dynamics simulations give insight into the conformational change, complex formation, and electron transfer pathway for cytochrome P450 reductase. Protein Sci. 2013, 22, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.C.; Ellis, J.; Moody, P.C.; Raven, E.L.; Roberts, G.C. Redox-linked domain movements in the catalytic cycle of cytochrome P450 reductase. Structure 2013, 21, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Ebrecht, A.C.; van der Bergh, N.; Harrison, S.T.; Smit, M.S.; Sewell, B.T.; Opperman, D.J. Biochemical and structural insights into the cytochrome P450 reductase from Candida tropicalis. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Campelo, D.; Lautier, T.; Urban, P.; Esteves, F.; Bozonnet, S.; Truan, G.; Kranendonk, M. The hinge segment of human NADPH-cytochrome P450 reductase in conformational switching: The critical role of ionic strength. Front. Pharmacol. 2017, 8, 755, Correction in Front. Pharmacol. 2018, 9, 175, doi:10.3389/fphar.2018.00175. [Google Scholar] [CrossRef]
- Laursen, T.; Jensen, K.; Møller, B.L. Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 132–138. [Google Scholar] [CrossRef]
- Iijima, M.; Ohnuki, J.; Sato, T.; Sugishima, M.; Takano, M. Coupling of Redox and Structural States in Cytochrome P450 Reductase Studied by Molecular Dynamics Simulation. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Campelo, D.; Esteves, F.; Palma, B.B.; Gomes, B.C.; Rueff, J.; Lautier, T.; Urban, P.; Truan, G.; Kranendonk, M. Probing the role of the hinge segment of cytochrome P450 oxidoreductase in the interaction with cytochrome P450. Int. J. Mol. Sci. 2018, 19, 3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Sligar, S.G. Modulation of the cytochrome P450 reductase redox potential by the phospholipid bilayer. Biochemistry 2009, 48, 12104–12112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Don, C.G.; Smieško, M. Microsecond MD simulations of human CYP2D6 wild-type and five allelic variants reveal mechanistic insights on the function. PLoS ONE 2018, 13, e0202534. [Google Scholar] [CrossRef] [PubMed]
- Gora, A.; Brezovsky, J.; Damborsky, J. Gates of enzymes. Chem. Rev. 2013, 113, 5871–5923. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Don, C.G.; Smieško, M. Molecular Dynamics Simulations Reveal Structural Differences among Allelic Variants of Membrane-Anchored Cytochrome P450 2D6. J. Chem. Inf. Model. 2018, 58, 1962–1975. [Google Scholar] [CrossRef]
- Berka, K.; Paloncýová, M.; Anzenbacher, P.; Otyepka, M. Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 2013, 117, 11556–11564. [Google Scholar] [CrossRef]
- Freeman, S.L.; Martel, A.; Devos, J.M.; Basran, J.; Raven, E.L.; Roberts, G.C. Solution structure of the cytochrome P450 reductase–cytochrome c complex determined by neutron scattering. J. Biol. Chem. 2018, 293, 5210–5219. [Google Scholar] [CrossRef] [Green Version]
- Orellana, L. Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 2019, 6, 117. [Google Scholar] [CrossRef]
- Fishelovitch, D.; Shaik, S.; Wolfson, H.J.; Nussinov, R. How does the reductase help to regulate the catalytic cycle of cytochrome P450 3A4 using the conserved water channel? J. Phys. Chem. B 2010, 114, 5964–5970. [Google Scholar] [CrossRef]
- Bridges, A.; Bruenke, L.; Chang, Y.T.; Vasker, I.A.; Loew, G.; Waskell, L. Identification of the Binding Site on Cytochrome P450 2B4 for Cytochrome b5 and Cytochrome P450 Reductase. J. Biol. Chem. 1998, 273, 17036–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šrejber, M.; Navrátilová, V.; Paloncýová, M.; Bazgier, V.; Berka, K.; Anzenbacher, P.; Otyepka, M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018, 183, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grunau, A.; Paine, M.; Munro, A.W.; Wolf, C.R.; Roberts, G.C.; Scrutton, N.S. Electron transfer in human cytochrome P450 reductase. Biochem. Soc. Trans. 2003, 31, 497–501. [Google Scholar] [CrossRef]
- Mukherjee, G.; Nandekar, P.; Wade, R. An electron transfer competent structural ensemble of membrane-bound cytochrome P450 1A1 and cytochrome P450 oxidoreductase. Commun. Biol. 2020, 4, 1–13. [Google Scholar]
- Kovrigina, E.A.; Pattengale, B.; Xia, C.; Galiakhmetov, A.R.; Huang, J.; Kim, J.J.P.; Kovrigin, E.L. Conformational States of Cytochrome P450 Oxidoreductase Evaluated by Förster Resonance Energy Transfer Using Ultrafast Transient Absorption Spectroscopy. Biochemistry 2016, 55, 5973–5976. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Hamdane, D.; Shen, A.L.; Choi, V.; Kasper, C.B.; Pearl, N.M.; Zhang, H.; Im, S.C.; Waskell, L.; Kim, J.J.P. Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 2011, 286, 16246–16260. [Google Scholar] [CrossRef] [Green Version]
- Sancho, J. Flavodoxins: Sequence, folding, binding, function and beyond. Cell. Mol. Life Sci. 2006, 63, 855–864. [Google Scholar] [CrossRef]
- Hoover, D.M.; Drennan, C.L.; Metzger, A.L.; Osborne, C.; Weber, C.H.; Pattridge, K.A.; Ludwig, M.L. Comparisons of wild-type and mutant flavodoxins from Anacystis nidulans. Structural determinants of the redox potentials. J. Mol. Biol. 1999, 294, 725–743. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Pattridge, K.A.; Metzger, A.L.; Dixon, M.M.; Eren, M.; Feng, Y.; Swenson, R.P. Control of oxidation-reduction potentials in flavodoxin from Clostridium beijerinckii: The role of conformation changes. Biochemistry 1997, 36, 1259–1280. [Google Scholar] [CrossRef]
- Rwere, F.; Xia, C.; Im, S.; Haque, M.M.; Stuehr, D.J.; Waskell, L.; Kim, J.J.P. Mutants of cytochrome P450 reductase lacking either gly-141 or gly-143 destabilize its FMN semiquinone. J. Biol. Chem. 2016, 291, 14639–14661. [Google Scholar] [CrossRef] [Green Version]
- Kandel, S.E.; Lampe, J.N. Role of protein-protein interactions in cytochrome P450-mediated drug metabolism and toxicity. Chem. Res. Toxicol. 2014, 27, 1474–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevrioukova, I.F.; Li, H.; Zhang, H.; Peterson, J.A.; Poulos, T.L.; Oulos, T.H.L.P. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritacco, I.; Saltalamacchia, A.; Spinello, A.; Ippoliti, E.; Magistrato, A. All-Atom simulations disclose how cytochrome reductase reshapes the substrate access/egress routes of its partner cyp450s. J. Phys. Chem. Lett. 2020, 11, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef] [PubMed]
- Leferink, N.G.; Pudney, C.R.; Brenner, S.; Heyes, D.J.; Eady, R.R.; Samar Hasnain, S.; Hay, S.; Rigby, S.E.; Scrutton, N.S. Gating mechanisms for biological electron transfer: Integrating structure with biophysics reveals the nature of redox control in cytochrome P450 reductase and copper-dependent nitrite reductase. FEBS Lett. 2012, 586, 578–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, A.W.; Leys, D.G.; McLean, K.J.; Marshall, K.R.; Ost, T.W.; Daff, S.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Moser, C.C.; et al. P450 BM3: The very model of a modern flavocytochrome. Trends Biochem. Sci. 2002, 27, 250–257. [Google Scholar] [CrossRef]
- Prade, E.; Mahajan, M.; Im, S.C.; Zhang, M.; Gentry, K.A.; Anantharamaiah, G.M.; Waskell, L.; Ramamoorthy, A. A Minimal Functional Complex of Cytochrome P450 and FBD of Cytochrome P450 Reductase in Nanodiscs. Angew. Chem. Int. Ed. 2018, 57, 8458–8462. [Google Scholar] [CrossRef]
- Sugishima, M.; Sato, H.; Higashimoto, Y.; Harada, J.; Wada, K.; Fukuyama, K.; Noguchi, M. Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, 2524–2529. [Google Scholar] [CrossRef] [Green Version]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta 2007, 1770, 390–401. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M. Ligand Pathways in Nuclear Receptors. J. Chem. Inf. Model. 2019, 59, 3100–3109. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, SC, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- LLC Schrödinger. Maestro Small-Molecule Drug Discovery Suite 2018-2; Schrodinger: New York, NY, USA, 2018. [Google Scholar]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.; Bonvin, A.M. A multidomain flexible docking approach to deal with large conformational changes in the modeling of biomolecular complexes. Structure 2011, 19, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Perez, T.E.; Mealey, K.L.; Grubb, T.L.; Greene, S.A.; Court, M.H. Tramadol metabolism to o-desmethyl tramadol (M1) and N-Desmethyl Tramadol (M2) by Dog Liver Microsomes: Species Comparison and Identification of Responsible Canine Cytochrome P450s. Drug Metab. Dispos. 2016, 44, 1963–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, C.; Oostenbrink, C.; Keizers, P.H.J.; Van Der Wijst, T.; Jongejan, A.; Vermeulen, N.P.E. Catalytic site prediction and virtual screening of cytochrome P450 2D6 substrates by consideration of water and rescoring in automated docking. J. Med. Chem. 2006, 49, 2417–2430. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sellner, M.; Fischer, A.; Don, C.G.; Smieško, M. Conformational Landscape of Cytochrome P450 Reductase Interactions. Int. J. Mol. Sci. 2021, 22, 1023. https://doi.org/10.3390/ijms22031023
Sellner M, Fischer A, Don CG, Smieško M. Conformational Landscape of Cytochrome P450 Reductase Interactions. International Journal of Molecular Sciences. 2021; 22(3):1023. https://doi.org/10.3390/ijms22031023
Chicago/Turabian StyleSellner, Manuel, André Fischer, Charleen G. Don, and Martin Smieško. 2021. "Conformational Landscape of Cytochrome P450 Reductase Interactions" International Journal of Molecular Sciences 22, no. 3: 1023. https://doi.org/10.3390/ijms22031023
APA StyleSellner, M., Fischer, A., Don, C. G., & Smieško, M. (2021). Conformational Landscape of Cytochrome P450 Reductase Interactions. International Journal of Molecular Sciences, 22(3), 1023. https://doi.org/10.3390/ijms22031023