
Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength


Abstract
:1. Introduction
2. Results
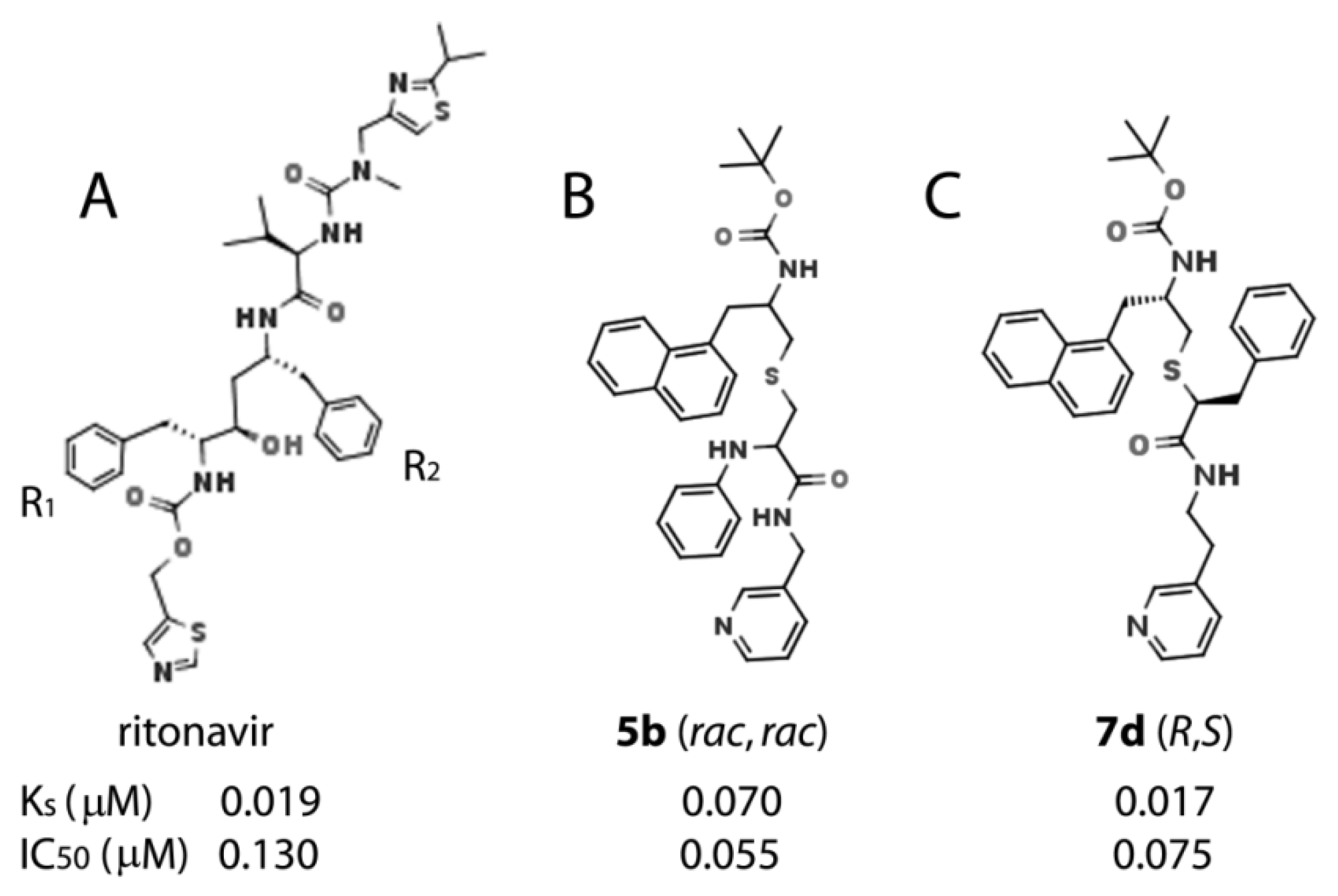
2.1. Rationale for Series V Analogues
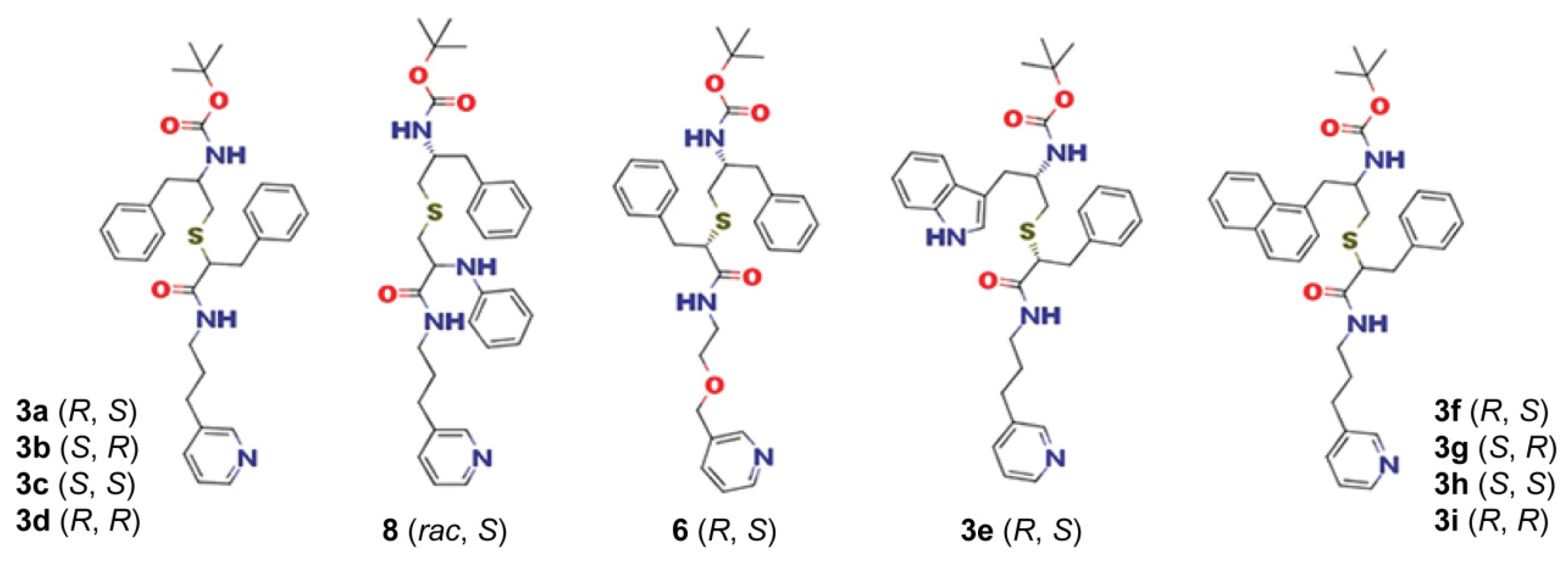
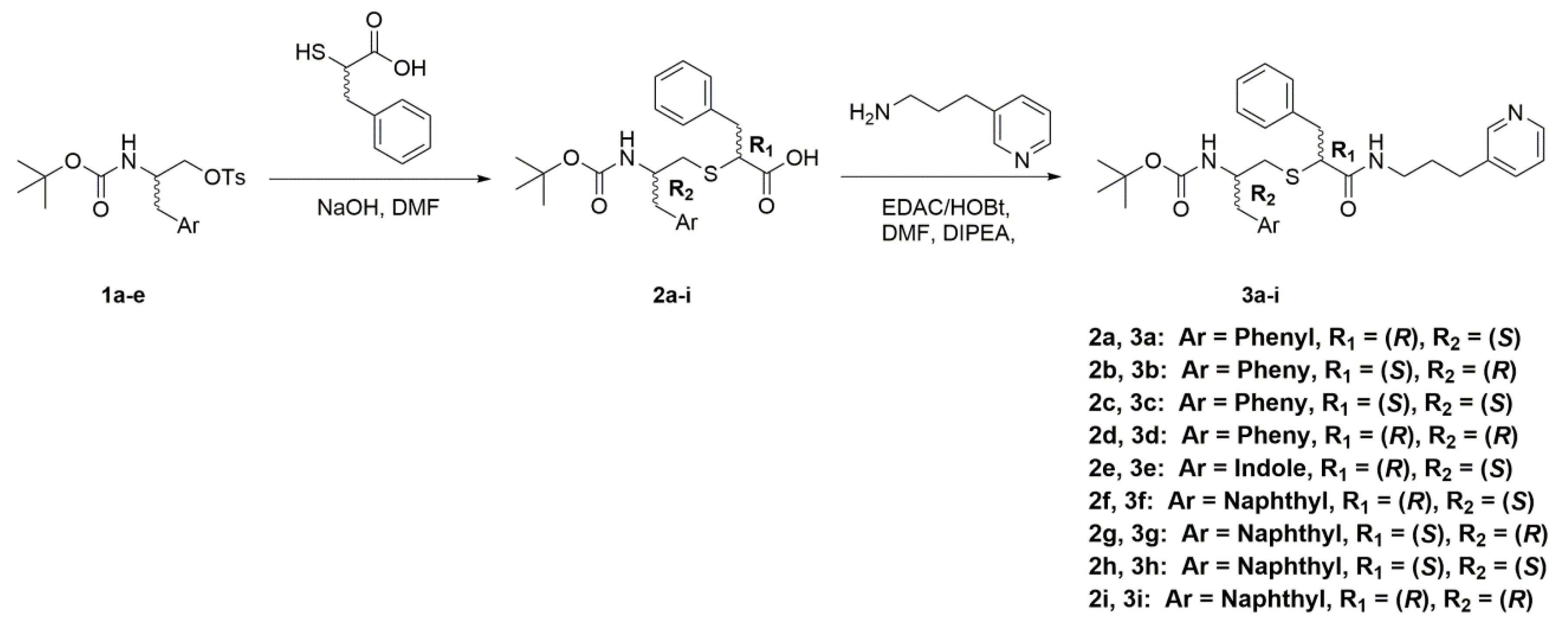
2.2. The 3a–d Analogues
2.2.1. Spectral and Biochemical Properties of 3a–d
2.2.2. Crystallization of 3a–d-Bound CYP3A4
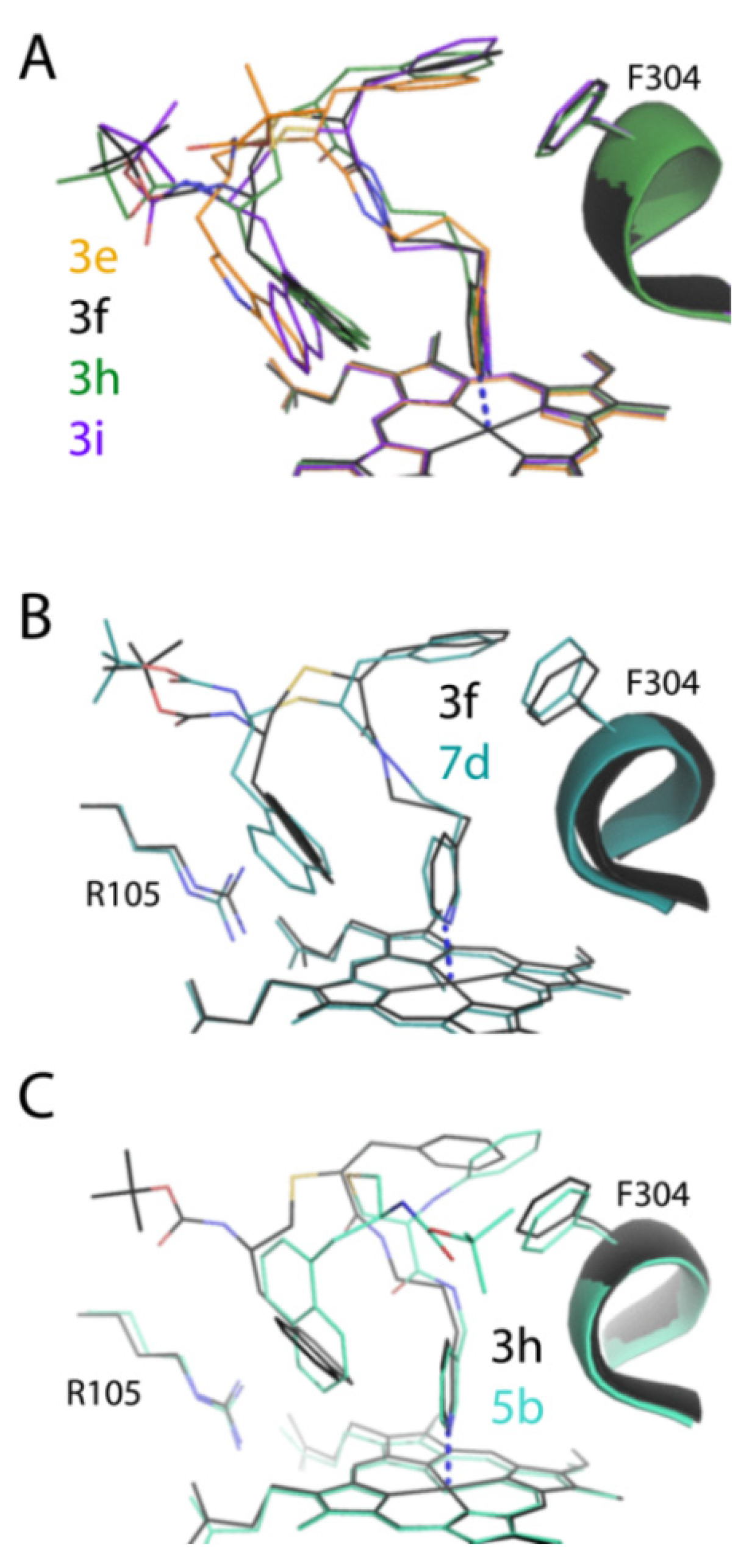
2.2.3. Compounds 3a–d Binding Modes
2.3. R1/R2 Spacer Extension Has No Beneficial Effect
2.4. Two-Atom Pyridyl-Linker Elongation Is Detrimental for the Binding to CYP3A4
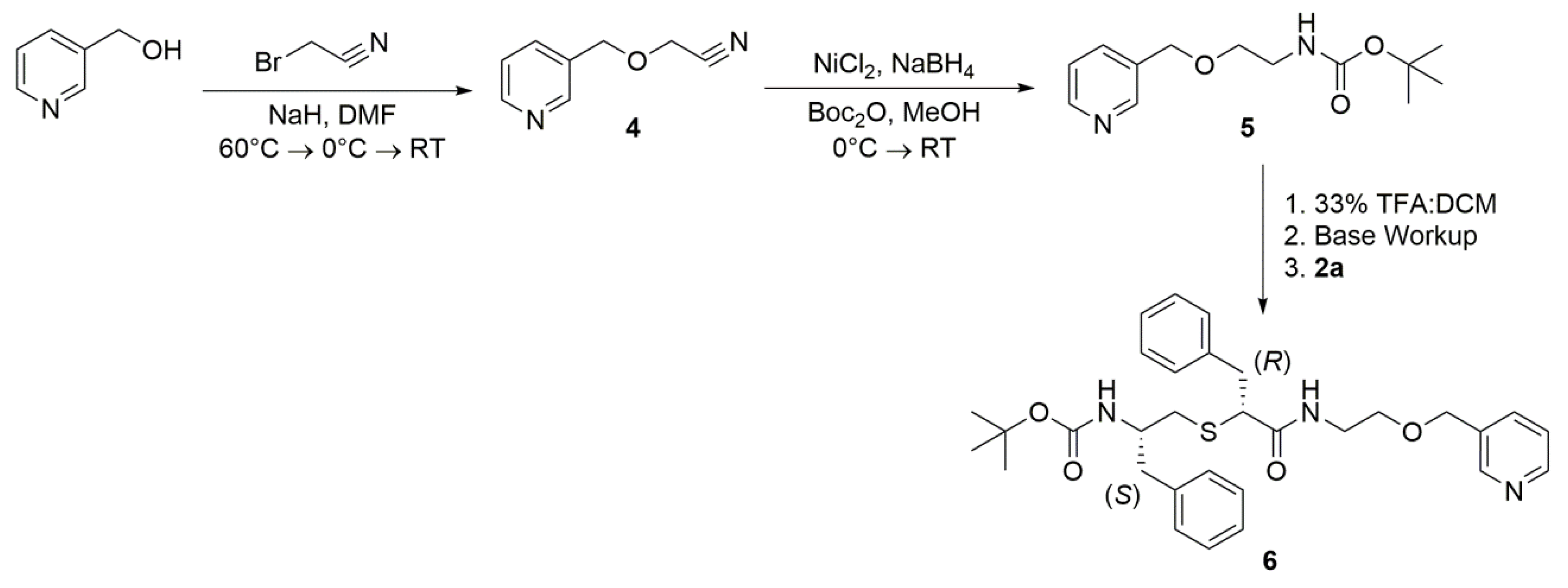
2.4.1. Synthetic Approaches for Linker Elongation
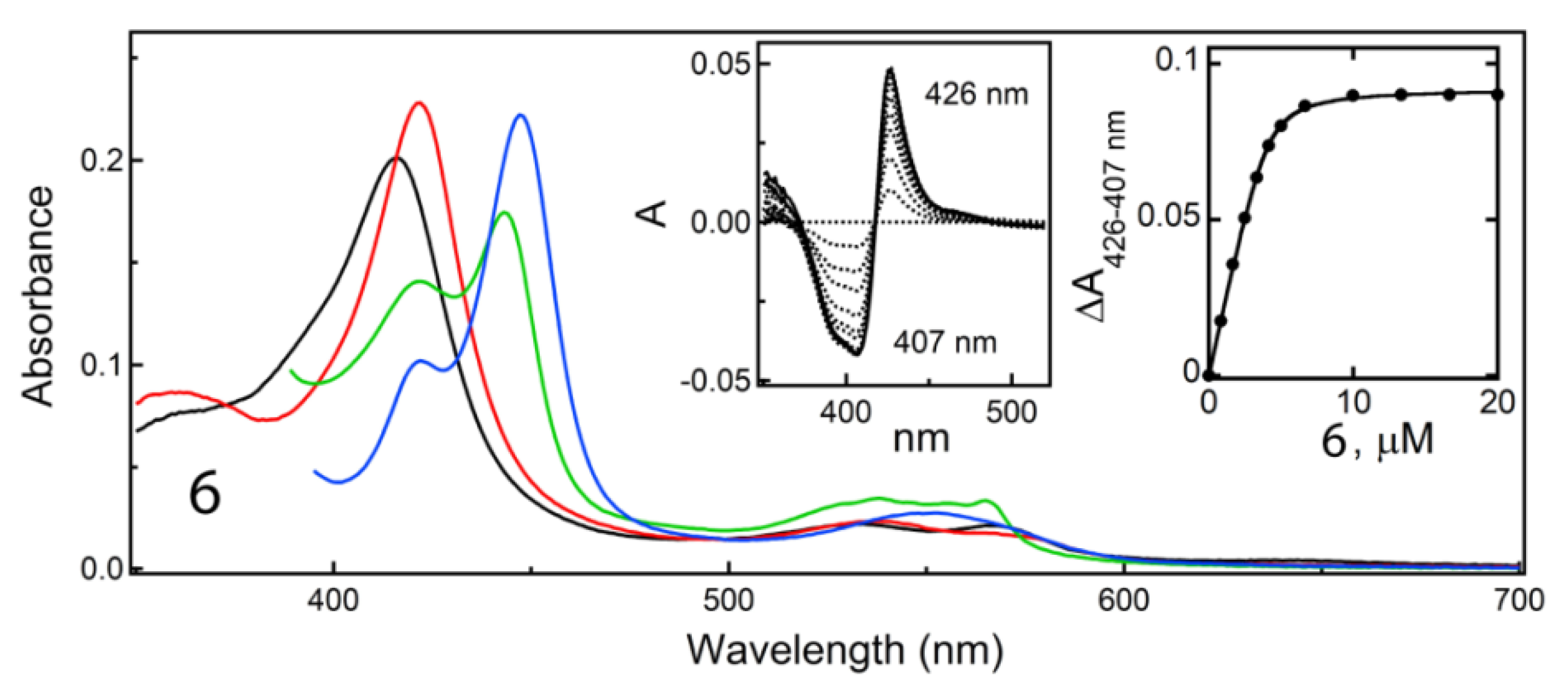
2.4.2. Properties of 6
2.5. The 3e–i Subseries
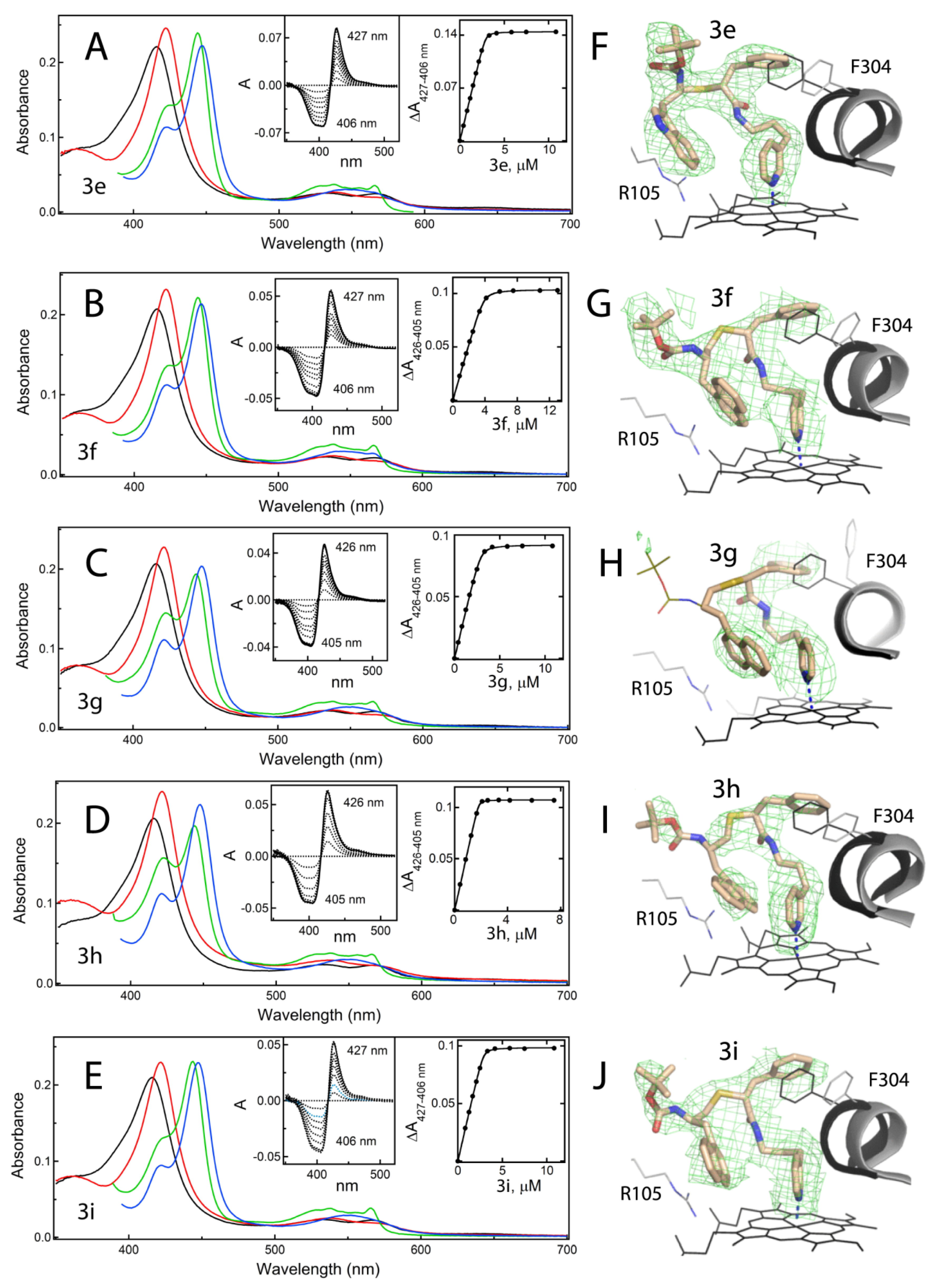
2.5.1. Spectral and Biochemical Properties of 3e–i
2.5.2. The 3e–i Binding Modes
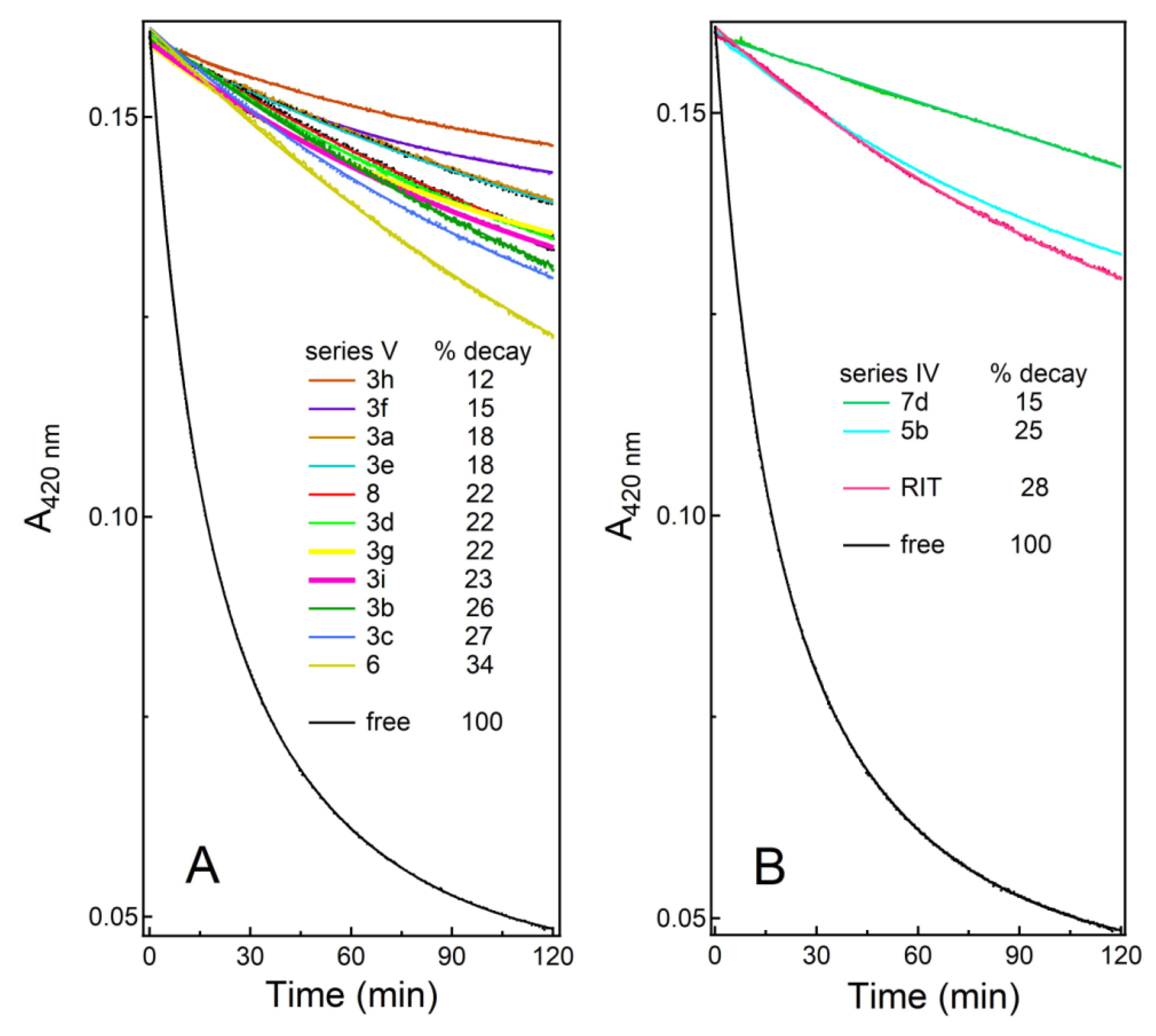
2.6. H2O2 Heme Accessibility Assessment
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP3A4 | Cytochrome P450 3A4 |
| BFC | 7-benzyloxy-4-(trifluoromethyl)coumarin |
| SAR | Structure–activity relationship |
| WT | Wild type |
References
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.P.; Peter Guengerich, F. Human cytochrome P450 enzymes 5-51 as targets of drugs and natural and environmental compounds: Mechanisms, induction, and inhibition—toxic effects and benefits. Drug Metab. Rev. 2018, 50, 256–342. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Shimada, T. Oxidation of toxic and carcinogenic chemicals by human cytochrome P-450 enzymes. Chem. Res. Toxicol. 1991, 4, 391–407. [Google Scholar] [CrossRef]
- Li, A.P.; Kaminski, D.L.; Rasmussen, A. Substrates of human hepatic cytochrome P450 3A4. Toxicology 1995, 104, 1–8. [Google Scholar] [CrossRef]
- Mehmood, Z.; Williamson, M.P.; Kelly, D.E.; Kelly, S.L. Metabolism of organochlorine pesticides: The role of human cytochrome P450 3A4. Chemosphere 1996, 33, 759–769. [Google Scholar] [CrossRef]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Liu, H.; Murray, B.; Callebaut, C.; Lee, M.S.; Hong, A.; Strickley, R.G.; Tsai, L.K.; Stray, K.M.; Wang, Y.; et al. Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med. Chem. Lett. 2010, 1, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef]
- Samuels, E.R.; Sevrioukova, I.F. An increase in side-group hydrophobicity largely improves the potency of ritonavir-like inhibitors of CYP3A4. Bioorg. Med. Chem. 2020, 28, 115349. [Google Scholar] [CrossRef]
- Brayer, S.W.; Reddy, K.R. Ritonavir-boosted protease inhibitor based therapy: A new strategy in chronic hepatitis C therapy. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, D.J. The ketoconazole legacy. Clin. Pharmacol. Drug Dev. 2014, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, D.J. Evidence-based choice of ritonavir as index CYP3A inhibitor in drug-drug interaction studies. J. Clin. Pharmacol. 2016, 56, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Desai, M.C. Pharmacokinetic enhancers for HIV drugs. Curr. Opin. Investig. Drugs 2009, 10, 775–786. [Google Scholar]
- Mathias, A.A.; German, P.; Murray, B.P.; Wei, L.; Jain, A.; West, S.; Warren, D.; Hui, J.; Kearney, B.P. Pharmacokinetics and pharmacodynamics of GS-9350: A novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010, 87, 322–329. [Google Scholar] [CrossRef]
- Hossain, M.A.; Tran, T.; Chen, T.; Mikus, G.; Greenblatt, D.J. Inhibition of human cytochromes P450 in vitro by ritonavir and cobicistat. J. Pharm. Pharmacol. 2017, 69, 1786–1793. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Interaction of human cytochrome P4503A4 with ritonavir analogs. Arch. Biochem. Biophys. 2012, 520, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Pyridine-substituted desoxyritonavir is a more potent cytochrome P450 3A4 inhibitor than ritonavir. J. Med. Chem. 2013, 56, 3733–3741. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr. Top. Med. Chem. 2014, 14, 1348–1355. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Chamberlin, A.R.; Poulos, T.L.; Sevrioukova, I.F. Structure-based inhibitor design for evaluation of a CYP3A4 pharmacophore model. J. Med. Chem. 2016, 59, 4210–4220. [Google Scholar] [CrossRef] [Green Version]
- Samuels, E.R.; Sevrioukova, I.F. Inhibition of human CYP3A4 by rationally designed ritonavir-like compounds: Impact and interplay of the side group functionalities. Mol. Pharm. 2018, 15, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Sevrioukova, I. Structure-activity relationships of rationally designed ritonavir analogs: Impact of side-group stereochemistry, head-group spacing, and backbone composition on the interaction with CYP3A4. Biochemistry 2019, 58, 2077–2087. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Section D 2017, 73, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Li, H.; Doud, E.H.; Chen, Y.; Choing, S.; Plaza, C.; Kelleher, N.L.; Poulos, T.L.; Silverman, R.B. Mechanism of Inactivation of Neuronal Nitric Oxide Synthase by (S)-2-Amino-5-(2-(methylthio)acetimidamido)pentanoic Acid. J. Am. Chem. Soc. 2015, 137, 5980–5989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettcher, A.; Pascal Furet, N.B.; Groell, J.-M.; Kallen, J.; Hergovich, L.J.; Masuya, K.; Mayr, L.; Vaupel, A. 3-Imidazolyl-Indoles for the Treatment of Proliferative Diseases. U.S. Patent 12,593,721, 27 March 2008. [Google Scholar]
- Becker, P.; Duhamel, T.; Stein, C.J.; Reiher, M.; Muniz, K. Cooperative light-activated iodine and photoredox catalysis for the amination of Csp3 -H bonds. Angew. Chem. 2017, 56, 8004–8008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szostak, M.; Sautier, B.; Spain, M.; Procter, D.J. Electron transfer reduction of nitriles using SmI2-Et3N-H2O: Synthetic utility and mechanism. Org. Lett. 2014, 16, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.B.L.; Liu, C.; Li, J.G. Ambiguity-Free Optical Tracking System. U.S. Patent 14,394,353, 12 April 2013. [Google Scholar]
- Guan, A.; Liu, C.; Chen, W.; Yang, F.; Xie, Y.; Zhang, J.; Li, Z.; Wang, M. Design, Synthesis, and Structure-Activity Relationship of New Pyrimidinamine Derivatives Containing an Aryloxy Pyridine Moiety. J. Agric. Food Chem. 2017, 65, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Itsuno, S.; Sakurai, Y.; Ito, K. Reduction of some functional groups with zirconium tetrachloride/sodium borohydride. Synth. Commun. 1988, 6, 995–996. [Google Scholar] [CrossRef]
- Caddick, S.; Haynes, A.K.D.; Judd, D.B.; Williams, M.R.V. Convenient synthesis of protected primary amines from nitriles. Tetrahedron Lett. 2000, 41, 3513–3516. [Google Scholar] [CrossRef]
- Caddick, S.; Judd, D.B.; Lewis, A.K.D.; Reich, M.T. A generic approach for the catalytic reduction of nitriles. Tetrahedron 2003, 59, 5417–5423. [Google Scholar] [CrossRef]
- Khurana, J.M.; Kukreja, G. Rapid reduction of nitriles to primary amines with nickel boride at ambient temperature. Synth. Commun. 2002, 32, 1265–1269. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikuleva, I.A.; Lapko, A.G.; Chashchin, V.L. Functional reconstitution of cytochrome P-450scc with hemin activated with Woodward’s reagent K. Formation of a hemeprotein cross-link. J. Biol. Chem. 1992, 267, 1438–1442. [Google Scholar] [CrossRef]
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.P.; et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. USA 1995, 92, 2484–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, D.J.; Harmatz, J.S. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450-3A in drug-drug interaction studies. Br. J. Clin. Pharmacol. 2015, 80, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koudriakova, T.; Iatsimirskaia, E.; Utkin, I.; Gangl, E.; Vouros, P.; Storozhuk, E.; Orza, D.; Marinina, J.; Gerber, N. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: Mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab. Dispos. 1998, 26, 552–561. [Google Scholar] [PubMed]
- von Moltke, L.L.; Durol, A.L.; Duan, S.X.; Greenblatt, D.J. Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: Comparison with ketoconazole. Eur. J. Clin. Pharmacol. 2000, 56, 259–261. [Google Scholar] [CrossRef]
- Ernest, C.S., 2nd; Hall, S.D.; Jones, D.R. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J. Pharmacol. Exp. Ther. 2005, 312, 583–591. [Google Scholar] [CrossRef]
- Lin, H.L.; D’Agostino, J.; Kenaan, C.; Calinski, D.; Hollenberg, P.F. The effect of ritonavir on human CYP2B6 catalytic activity: Heme modification contributes to the mechanism-based inactivation of CYP2B6 and CYP3A4 by ritonavir. Drug Metab. Dispos. 2013, 41, 1813–1824. [Google Scholar] [CrossRef]
- Rock, B.M.; Hengel, S.M.; Rock, D.A.; Wienkers, L.C.; Kunze, K.L. Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol. Pharmacol. 2014, 86, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Caciolla, J.; Spinello, A.; Martini, S.; Bisi, A.; Zaffaroni, N.; Gobbi, S.; Magistrato, A. Targeting Orthosteric and Allosteric Pockets of Aromatase via Dual-Mode Novel Azole Inhibitors. ACS Med. Chem. Lett. 2020, 11, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.; Sevrioukova, I. Direct synthesis of alpha-thio aromatic acids from aromatic amino acids. Tetrahedron Lett. 2018, 59, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F. High-level production and properties of the cysteine-depleted cytochrome P450 3A4. Biochemistry 2017, 56, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax (nm) | A421/417 a | ΔAmax b | Ks c | IC50 d | IC50/Ks | ΔTm e | kfastf | kET g |
|---|---|---|---|---|---|---|---|---|---|
| Ferric/ferrous | μM | μM | °C | s−1 | s−1 | ||||
| Pyridyl-propyl linker | |||||||||
| 4-atom R1(phenyl)-R2(phenyl) spacer | |||||||||
| 3a (R, S) | 422/444 | 1.13 | 122% | 0.015 ± 0.003 | 0.16 ± 0.02 | 10.7 | 8.1 | 12.5 (36%) | 0.011 (98%) |
| 3b (S, R) | 421/443 | 1.08 | 111% | 0.029 ± 0.004 | 0.30 ± 0.02 | 10.3 | 5.3 | 9.5 (35%) | 0.014 (87%) |
| 3c (S, S) | 422/444 | 1.09 | 111% | 0.026 ± 0.002 | 0.31 ± 0.04 | 11.9 | 5.8 | 9.5 (33%) | 0.012 (77%) |
| 0.012 (77%) | 422/444 | 1.10 | 114% | 0.013 ± 0.005 | 0.21 ± 0.02 | 20.0 | 5.6 | 12.0 (38%) | 0.013 (95%) |
| 5-atom R1(phenyl)-R2(phenyl) spacer | |||||||||
| 8 (rac, S) | 422/444 | 1.10 | 114% | 0.028 ± 0.004 | 0.33 ± 0.03 | 7.5 | 5.0 | 16.2 (30%) | 0.018 (58%) |
| Pyridyl-mehtoxyethyl (butyl-like) linker | |||||||||
| 4-atom R1(phenyl)-R2(phenyl) spacer | |||||||||
| 6 (R, S) | 422/443 | 1.07 | 113% | 0.213 ± 0.009 | 0.73 ± 0.06 | 3.4 | 3.0 | 9.5 (29%) | 0.017 (65%) |
| Pyridyl-propyl linker | |||||||||
| 4-atom R1(phenyl)-R2(indol) spacer | |||||||||
| 3e (R, S) | 422/444 | 1.11 | 125% | 0.019 ± 0.002 | 0.15 ± 0.03 | 7.9 | 7.8 | 20.1 (24%) | 0.020 (85%) |
| Pyridyl-propyl linker | |||||||||
| 4-atom R1(phenyl)-R2(naphthalene) spacer | |||||||||
| 3f (R, S) | 422/444 | 1.12 | 125% | 0.015 ± 0.001 | 0.16 ± 0.03 | 10.7 | 7.9 | 19.3 (28%) | 0.011 (94%) |
| 3g (S, R) | 422/444 | 1.09 | 111% | 0.024 ± 0.006 | 0.30 ± 0.06 | 12.5 | 4.9 | 10.8 (32%) | 0.011 (75%) |
| 3h (S, S) | 422/444 | 1.09 | 112% | 0.007 ± 0.001 | 0.09 ± 0.01 | 12.9 | 5.9 | 16.1 (24%) | 0.017 (72%) |
| 3i (R, R) | 422/444 | 1.10 | 114% | 0.018 ± 0.003 | 0.15 ± 0.02 | 8.3 | 5.5 | 12.3 (24%) | 0.021 (97%) |
| Compound | Fe–N Bond | Pyridine ring | I-helix | H-Bond | Pyridine–R2 Ring | F304–R1 Ring | Boc-Group | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Distance (Å) Angle (°) a | Rotation (°) b | Shift (Å) c | with S119 (Å) d | Angle and Overlap | Angle and Overlap | Contacts | ||||
| 3a (R, S) mol A | 2.11 | 3 | 30 | 2.04–2.18 | 2.82 | 42° | full | 50° | partial | 108, 211, 213 |
| 3b (S, R) | 2.20 | 15 | 17 | 1.09–1.76 | 3.25 | 46° | half e | 65° | partial e | disordered |
| 3c (S, S) | 2.18 | 7 | 12 | 1.23–1.76 | 3.08 | 35° | partial | 36° | half | 105, 106, 108, 374 |
| 3d (R, R) mol A | 2.18 | 0 | 37 | 2.11–2.18 | 2.48 | 5° | full | 50° | half | 57, 108, 211, 213, 482 |
| 8 (R, S) | 2.11 | 3 | 32 | 1.77–1.97 | 3.15 f | 25° | half | 20° | full | disordered |
| 3e (R, S) mol A | 2.08 | 2 | 37 | 0.81–1.34 | 2.30 | 33° | full | 50° | half | 108, 211, 213 |
| 3f (R, S) mol A | 2.07 | 0 | 33 | 1.90–2.11 | 2.41 | 45° | half | 40° | half | 105–108 |
| 3g (S, R) | 2.23 | 6 | 10 | 0.47–0.42 | - | 30° | half | 85° | none | disordered |
| 3h (S, S) mol A | 2.07 | 3 | 35 | 1.98–2.21 | 2.35 | 45° | full | 40° | half | 105–108 |
| 3i (R, R) mol A | 2.09 | 4 | 30 | 1.96–2.14 | 2.30 | 20° | full | 30° | half | 105–108, 374 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samuels, E.R.; Sevrioukova, I.F. Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength. Int. J. Mol. Sci. 2021, 22, 852. https://doi.org/10.3390/ijms22020852
Samuels ER, Sevrioukova IF. Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength. International Journal of Molecular Sciences. 2021; 22(2):852. https://doi.org/10.3390/ijms22020852
Chicago/Turabian StyleSamuels, Eric R., and Irina F. Sevrioukova. 2021. "Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength" International Journal of Molecular Sciences 22, no. 2: 852. https://doi.org/10.3390/ijms22020852
APA StyleSamuels, E. R., & Sevrioukova, I. F. (2021). Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength. International Journal of Molecular Sciences, 22(2), 852. https://doi.org/10.3390/ijms22020852