
Inflammatory Depression—Mechanisms and Non-Pharmacological Interventions


Abstract
:1. Introduction
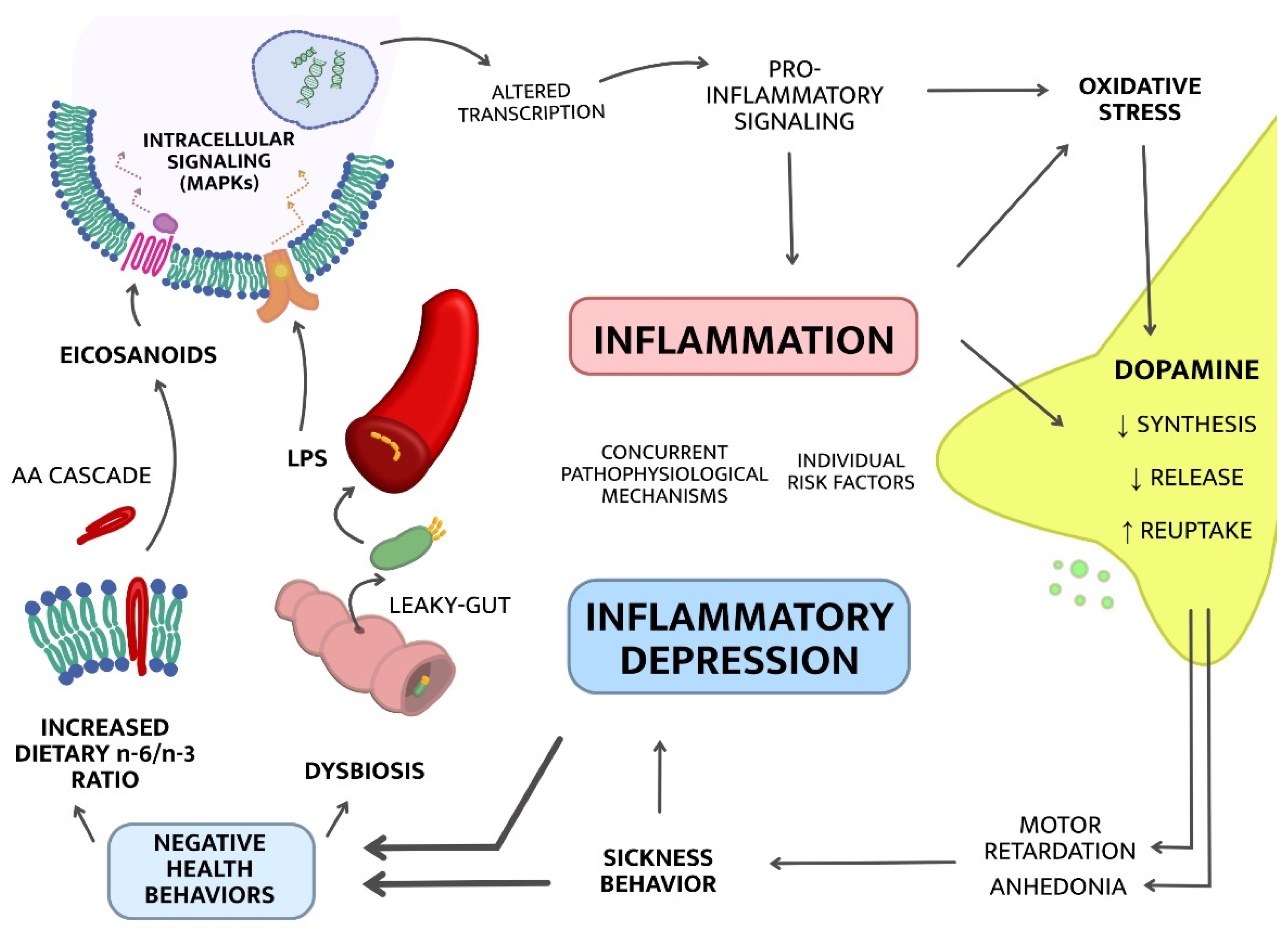
2. Mechanisms of “Inflammatory Depression”
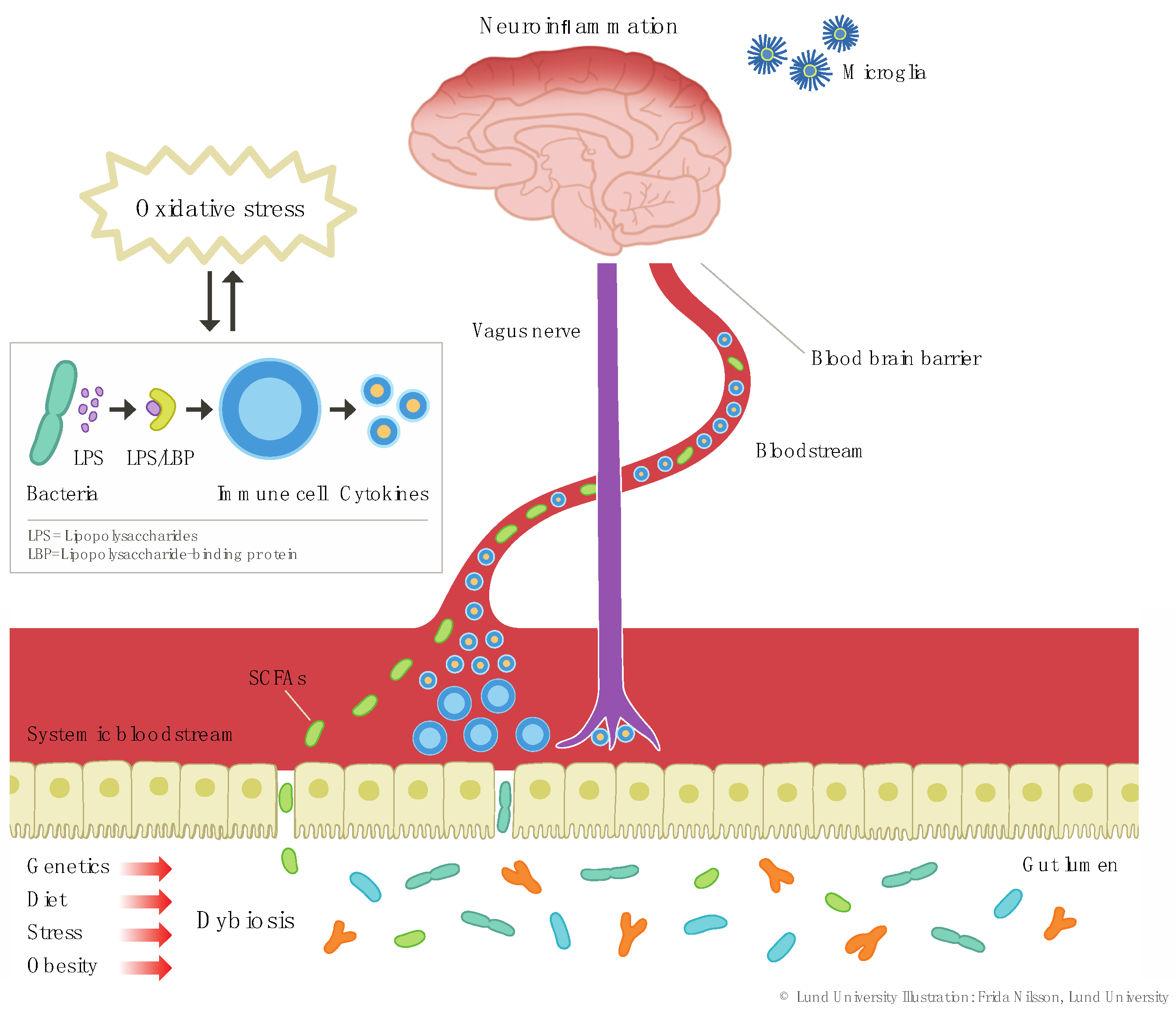
2.1. Upstream Mechanisms; from Lifestyle to Inflammation
2.1.1. Dysbiosis
2.1.2. Increased Dietary Omega-6/Omega-3 Ratio
2.2. Downstream Mechanisms; from Inflammation to Symptoms
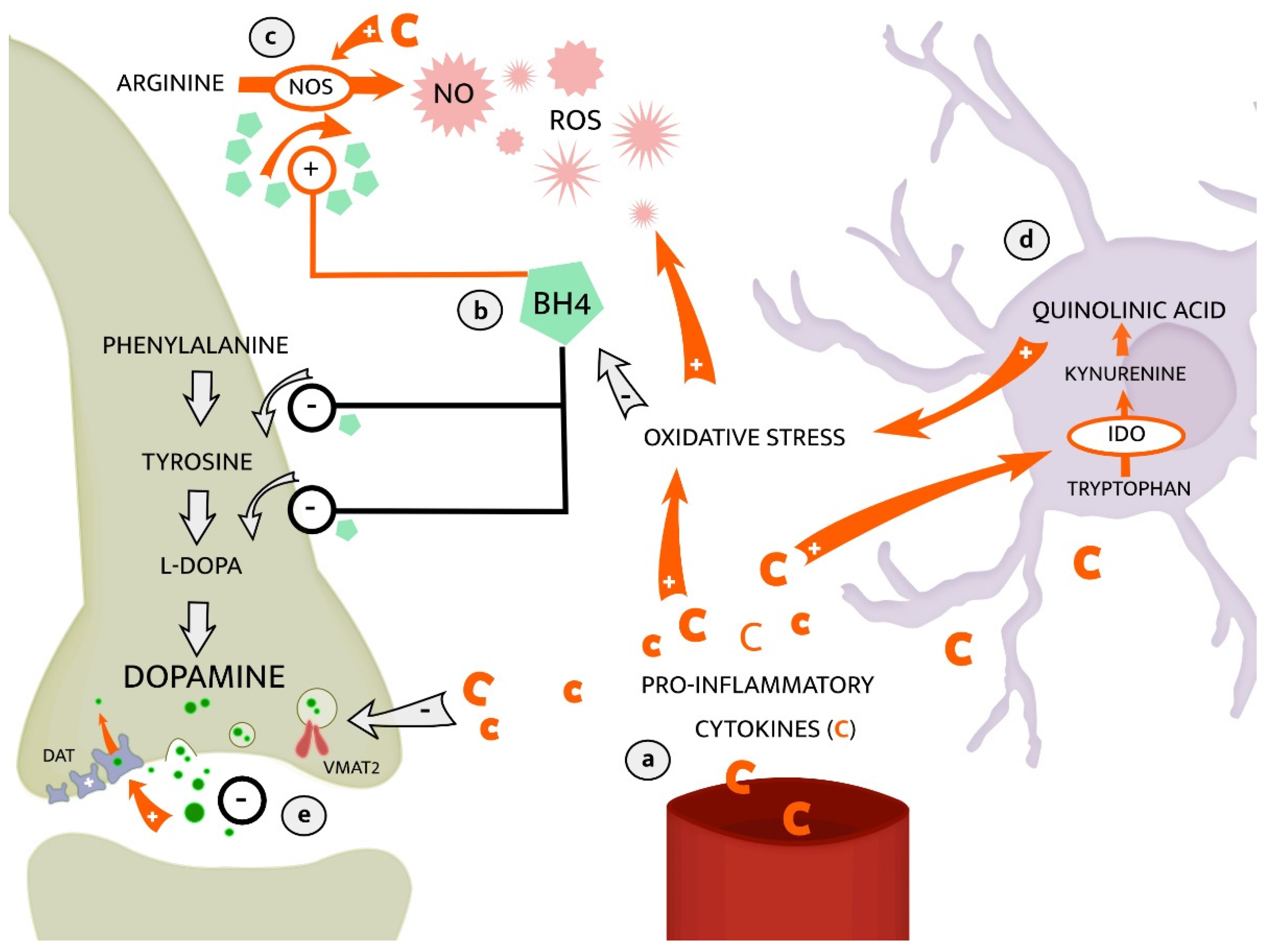
2.2.1. The Kynurenine Pathway of Tryptophan Metabolism
2.2.2. Dopaminergic Neurotransmission
3. Therapeutic Implications
3.1. Omega-3 Fatty Acids as a Potential Treatment of Inflammatory Depression
3.2. Dysbiosis, Inflammation, and Depression—Are Probiotics Efficacious in Inflammatory Depression?
3.3. Is Exercise Efficacious in Inflammatory Depression?
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, D. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, D. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol. Psychiatry 2009, 66, 287–292. [Google Scholar] [CrossRef]
- Felger, J.C. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haroon, E. Conceptual convergence: Increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol. Psychiatry 2016, 21, 1351–1357. [Google Scholar] [CrossRef]
- Lindqvist, D. Cerebrospinal fluid inflammatory markers in Parkinson’s disease—Associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Raison, C.L.; Miller, A.H. Is depression an inflammatory disorder? Curr. Psychiatry Rep. 2011, 13, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Haroon, E.; Felger, J.C. Therapeutic Implications of Brain-Immune Interactions: Treatment in Translation. Neuropsychopharmacology 2017, 42, 334–359. [Google Scholar] [CrossRef] [PubMed]
- Jokela, M. Inflammation and Specific Symptoms of Depression. JAMA Psychiatry 2016, 73, 87–88. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Trivedi, M.H. Personalized Antidepressant Selection and Pathway to Novel Treatments: Clinical Utility of Targeting Inflammation. Int. J. Mol. Sci. 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler-Forsberg, O. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: Meta-analysis of clinical trials. Acta Psychiatr. Scand. 2019, 139, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Köhler, O. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Derry, H.M.; Fagundes, C.P. Inflammation: Depression fans the flames and feasts on the heat. Am. J. Psychiatry 2015, 172, 1075–1091. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Chan, Y.K.; Estaki, M.; Gibson, D.L. Clinical consequences of diet-induced dysbiosis. Ann. Nutr. Metab. 2013, 63, 28–40. [Google Scholar] [CrossRef]
- Sohail, M.U. Impact of Physical Exercise on Gut Microbiome, Inflammation, and the Pathobiology of Metabolic Disorders. Rev. Diabet. Stud. 2019, 15, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.F. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Savin, Z. Smoking and the intestinal microbiome. Arch. Microbiol. 2018, 200, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Liang, S. Recognizing Depression from the Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2018, 19, 1592. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.R. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, R.R. Structure and function of lipopolysaccharide binding protein. Science 1990, 249, 1429–1431. [Google Scholar] [CrossRef]
- Maes, M. Increased IgA and IgM responses against gut commensals in chronic depression: Further evidence for increased bacterial translocation or leaky gut. J. Affect. Disord. 2012, 141, 55–62. [Google Scholar] [CrossRef]
- Li, M. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Braniste, V. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawan, E. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef]
- Erny, D. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Thion, M.S. Microbiome Influences Prenatal and Adult Microglia in a Sex.-Specific Manner. Cell 2018, 172, 500–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Kelly, J.R. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wood, T.K.; Lee, J. Roles of indole as an interspecies and interkingdom signaling molecule. Trends Microbiol. 2015, 23, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. 2018, 32, 6681–6693. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, A.N.; Tingö, L.; Brummer, R.J. The Potential Effects of Probiotics and ω-3 Fatty Acids on Chronic Low-Grade Inflammation. Nutrients 2020, 12, 2402. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef]
- Wall, R. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Messamore, E. Polyunsaturated fatty acids and recurrent mood disorders: Phenomenology, mechanisms, and clinical application. Prog. Lipid Res. 2017, 66, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br. J. Clin. Pharmacol. 2013, 75, 645–662. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Tilley, S.L.; Coffman, T.M.; Koller, B.H. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest. 2001, 108, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bavaresco, D.V. Evaluation of the arachidonic acid pathway in bipolar disorder: A systematic review. Mol. Biol. Rep. 2020, 47, 8209–8217. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Mode of action of mood stabilizers: Is the arachidonic acid cascade a common target? Mol. Psychiatry 2008, 13, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Su, K.P. Phospholipase A2 and cyclooxygenase 2 genes influence the risk of interferon-alpha-induced depression by regulating polyunsaturated fatty acids levels. Biol. Psychiatry 2010, 67, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Noponen, M. Elevated PLA2 activity in schizophrenics and other psychiatric patients. Biol. Psychiatry 1993, 34, 641–649. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Bosetti, F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch. Gen. Psychiatry 2002, 59, 592–596. [Google Scholar] [CrossRef]
- Rintala, J. 85 kDa cytosolic phospholipase A2 is a target for chronic lithium in rat brain. Neuroreport 1999, 10, 3887–3890. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Savitz, J. Role of Kynurenine Metabolism Pathway Activation in Major Depressive Disorders. Curr. Top Behav. Neurosci. 2017, 31, 249–267. [Google Scholar] [PubMed]
- Schwarcz, R. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, C. Effect of immune activation on the kynurenine pathway and depression symptoms—A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2020, 118, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Ogyu, K. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Laugeray, A. Peripheral and cerebral metabolic abnormalities of the tryptophan-kynurenine pathway in a murine model of major depression. Behav. Brain Res. 2010, 210, 84–91. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Stone, T.W.; Darlington, L.G. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br. J. Pharmacol. 2013, 169, 1211–1227. [Google Scholar] [CrossRef]
- Erhardt, S. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef]
- O’Connor, J.C. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2, 3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.K. Association of a polymorphism in the indoleamine- 2,3-dioxygenase gene and interferon-α-induced depression in patients with chronic hepatitis C. Mol. Psychiatry 2012, 17, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilmas, C. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Alzarea, S.; Rahman, S. Alpha-7 nicotinic receptor allosteric modulator PNU120596 prevents lipopolysaccharide-induced anxiety, cognitive deficit and depression-like behaviors in mice. Behav. Brain Res. 2019, 366, 19–28. [Google Scholar] [CrossRef]
- Quak, J. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology 2014, 45, 202–210. [Google Scholar] [CrossRef]
- Haroon, E. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef]
- Treadway, M.T.; Cooper, J.A.; Miller, A.H. Can’t or Won’t? Immunometabolic Constraints on Dopaminergic Drive. Trends Cogn. Sci. 2019, 23, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Treadway, M.T. Inflammation Effects on Motivation and Motor Activity: Role of Dopamine. Neuropsychopharmacology 2017, 42, 216–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuron, L. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch. Gen. Psychiatry. 2012, 69, 1044–1053. [Google Scholar] [CrossRef]
- Felger, J.C. Chronic interferon-α decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 2013, 38, 2179–2187. [Google Scholar] [CrossRef]
- Rawdin, B.J. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 2013, 31, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Kitagami, T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: Role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003, 978, 104–114. [Google Scholar] [CrossRef]
- Felger, J.C. Tyrosine metabolism during interferon-alpha administration: Association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 2013, 31, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Inserra, A. Neuroimmunomodulation in Major Depressive Disorder: Focus on Caspase 1, Inducible Nitric Oxide Synthase, and Interferon-Gamma. Mol. Neurobiol. 2019, 56, 4288–4305. [Google Scholar] [CrossRef] [Green Version]
- Beck, I.M. Crosstalk in inflammation: The interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [PubMed]
- Morón, J.A. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J. Neurosci. 2003, 23, 8480–8488. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Huang, S.Y.; Su, K.P. A meta-analytic review of polyunsaturated fatty acid compositions in patients with depression. Biol. Psychiatry 2010, 68, 140–147. [Google Scholar] [CrossRef]
- Appleton, K.M. Omega-3 fatty acids for depression in adults. Cochrane Database Syst. Rev. 2015, 11, CD004692. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, B. Efficacy of omega-3 highly unsaturated fatty acids in the treatment of depression. Br. J. Psychiatry 2016, 209, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.D. High-dose omega-3 polyunsaturated fatty acid supplementation might be more superior than low-dose for major depressive disorder in early therapy period: A network meta-analysis. BMC Psychiatry 2020, 20, 248. [Google Scholar] [CrossRef] [PubMed]
- Guu, T.W. International Society for Nutritional Psychiatry Research Practice Guidelines for Omega-3 Fatty Acids in the Treatment of Major Depressive Disorder. Psychother. Psychosom. 2019, 88, 263–273. [Google Scholar] [CrossRef]
- Mocking, R.J. Meta-analysis and meta-regression of omega-3 polyunsaturated fatty acid supplementation for major depressive disorder. Transl. Psychiatry 2016, 6, e756. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z. Fish. Oil Prevents Lipopolysaccharide-Induced Depressive-Like Behavior by Inhibiting Neuroinflammation. Mol. Neurobiol. 2017, 54, 7327–7334. [Google Scholar] [CrossRef]
- Su, K.P. Omega-3 fatty acids in the prevention of interferon-alpha-induced depression: Results from a randomized, controlled trial. Biol. Psychiatry 2014, 76, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, M.H. Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: A proof of concept study. Mol. Psychiatry 2016, 21, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Pariante, C.M. Trial failures of anti-inflammatory drugs in depression. Lancet Psychiatry 2020, 7, 837. [Google Scholar] [CrossRef]
- Bharwani, A. Structural & functional consequences of chronic psychosocial stress on the microbiome & host. Psychoneuroendocrinology 2016, 63, 217–227. [Google Scholar]
- Bailey, M.T. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.M. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizawa, E. Possible association of Bifidobacterium and Lactobacillus in the gut microbiota of patients with major depressive disorder. J. Affect Disord. 2016, 202, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Valles-Colomer, M. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef]
- Logan, A.C.; Katzman, M. Major depressive disorder: Probiotics may be an adjuvant therapy. Med. Hypotheses 2005, 64, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.X. A meta-analysis of the use of probiotics to alleviate depressive symptoms. J. Affect Disord. 2018, 228, 13–19. [Google Scholar] [CrossRef]
- Thomas, C.M. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS ONE 2012, 7, e31951. [Google Scholar] [CrossRef] [Green Version]
- Karimi, S. Lactobacillus reuteri strains protect epithelial barrier integrity of IPEC-J2 monolayers from the detrimental effect of enterotoxigenic Escherichia coli. Physiol. Rep. 2018, 6, e13514. [Google Scholar] [CrossRef] [Green Version]
- Ahrne, S.; Hagslatt, M.L. Effect of lactobacilli on paracellular permeability in the gut. Nutrients 2011, 3, 104–117. [Google Scholar] [CrossRef]
- Qiao, Y. Alterations of the gut microbiota in high-fat diet mice is strongly linked to oxidative stress. Appl. Microbiol. Biotechnol. 2013, 97, 1689–1697. [Google Scholar] [CrossRef]
- Bravo, J.A. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J. Probiotics normalize the gut-brain-microbiota axis in immunodeficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G793–G802. [Google Scholar] [CrossRef]
- Dickerson, F. Adjunctive probiotic microorganisms to prevent rehospitalization in patients with acute mania: A randomized controlled trial. Bipolar Disord. 2018, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Brenner, L.A. Evaluation of an Immunomodulatory Probiotic Intervention for Veterans With Co-occurring Mild Traumatic Brain Injury and Posttraumatic Stress Disorder: A Pilot Study. Front. Neurol. 2020, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Akkasheh, G. Clinical and metabolic response to probiotic administration in patients with major depressive disorder: A randomized, double-blind, placebo-controlled trial. Nutrition 2016, 32, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Majeed, M. MTCC 5856 for the management of major depression with irritable bowel syndrome: A randomised, double-blind, placebo controlled, multi-centre, pilot clinical study. Food Nutr. Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talarowska, M. Impact of oxidative/nitrosative stress and inflammation on cognitive functions in patients with recurrent depressive disorders. Med. Sci. Monit. 2014, 20, 110–115. [Google Scholar]
- Hansberry, D.R. Fecal Myeloperoxidase as a Biomarker for Inflammatory Bowel Disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Sanchez, M.I. Probiotic Bifidobacterium longum NCC3001 Reduces Depression Scores and Alters Brain Activity: A Pilot Study in Patients With Irritable Bowel Syndrome. Gastroenterology 2017, 153, 448–459. [Google Scholar] [CrossRef]
- Kazemi, A. Effect of probiotic and prebiotic vs placebo on psychological outcomes in patients with major depressive disorder: A randomized clinical trial. Clin. Nutr. 2019, 38, 522–528. [Google Scholar] [CrossRef]
- Schuch, F.B. Exercise as a treatment for depression: A meta-analysis adjusting for publication bias. J. Psychiatr. Res. 2016, 77, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morres, I.D. Aerobic exercise for adult patients with major depressive disorder in mental health services: A systematic review and meta-analysis. Depress. Anxiety 2019, 36, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rethorst, C.D.; Wipfli, B.M.; Landers, D.M. The antidepressive effects of exercise: A meta-analysis of randomized trials. Sports Med. 2009, 39, 491–511. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Depression in Adults: Recognition and Management; National Institute for Health and Care Excellence: London, UK, 2009. [Google Scholar]
- Krogh, J. Exercise for patients with major depression: A systematic review with meta-analysis and trial sequential analysis. BMJ Open 2017, 7, e014820. [Google Scholar] [CrossRef]
- Phillips, C.; Fahimi, A. Immune and Neuroprotective Effects of Physical Activity on the Brain in Depression. Front. Neurosci. 2018, 12, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Significance of gastrointestinal tract in the therapeutic mechanisms of exercise in depression: Synchronism between brain and intestine through GBA. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 103, 109971. [Google Scholar] [CrossRef]
- Rethorst, C.D. Pro-inflammatory cytokines as predictors of antidepressant effects of exercise in major depressive disorder. Mol. Psychiatry 2013, 18, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Lavebratt, C. Interleukin-6 and depressive symptom severity in response to physical exercise. Psychiatry Res. 2017, 252, 270–276. [Google Scholar] [CrossRef]
- Walsh, N.P. Position statement. Part. two: Maintaining immune health. Exerc. Immunol. Rev. 2011, 17, 64–103. [Google Scholar] [PubMed]
- Pedersen, B.K. The diseasome of physical inactivity—And the role of myokines in muscle—Fat cross talk. J. Physiol. 2009, 587, 5559–5568. [Google Scholar] [CrossRef]
- Ignácio, Z.M. Physical Exercise and Neuroinflammation in Major Depressive Disorder. Mol. Neurobiol. 2019, 56, 8323–8335. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M. Does PGC1α/FNDC5/BDNF Elicit the Beneficial Effects of Exercise on Neurodegenerative Disorders? Neuromol. Med 2016, 18, 1–15. [Google Scholar] [CrossRef]
- Rudzki, L. Probiotic Lactobacillus Plantarum 299v decreases kynurenine concentration and improves cognitive functions in patients with major depression: A double-blind, randomized, placebo controlled study. Psychoneuroendocrinology 2019, 100, 213–222. [Google Scholar] [CrossRef]
- Uher, R. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am. J. Psychiatry 2014, 171, 1278–1286. [Google Scholar] [CrossRef]
- Jha, M.K. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology 2017, 78, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N.V. Depression severity is associated with increased inflammation in veterans with peripheral artery disease. Vasc. Med. 2018, 23, 445–453. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Sample | Study Type | Biomarkers | Main Findings | Notes and Limitations |
|---|---|---|---|---|---|
| Rapaport et al., 2016 [85] | N = 155, all MDD and unmedicated | RCT, 8 with tx: EPA, DHA, or placebo | IL-1, IL-6, CRP, leptin, adiponectin | No overall tx effects. Subjects with high inflammation improved more on EPA than placebo | Evidence for dose-response effect with increasing EPA-placebo separation with increasing number of elevated inflammation markers. Proof-of-concept study with post hoc analyses in need of replication. |
| Akkasheh et al., 2016 [105] | N = 40, all MDD and unmedicated | RCT, 8 with tx: probiotics or placebo | Hs-CRP, p-glucose, total antioxidant capacity, lipids and more | Probiotic tx associated with significant improvement in depressive symptoms and decreased insulin and CRP levels | Probiotic strain not specified. Small sample size, in need of replication to study effect on inflammatory markers and lipids |
| Rudzki et al., 2018 [125] | N = 79, all MDD and SSRI-tx | RCT, 8 with tx: probiotics or placebo | IL-6, IL-1β, TNF-α, CRP, cortisol, TSH, kynurenine pathway-metabolites | Probiotic tx not associated with significant antidepressant or anti-inflammatory effect. Decrease in kynurenine concentration and improvement of cognitive function in the probiotic group | To our knowledge, 1st evidence for the effect of a probiotic intervention on the kynurenine pathway. |
| Rethorst et al., 2013 [118] | N = 105, all MDD and SSRI-tx. | Subjects randomly assigned to 12-week: low or high dose exercise intervention. Raters blinded to group assignment | IFN-γ, IL-6, IL-1β, TNF-α | TNF-α levels at baseline predicted better outcome. Delta depressive symptoms significantly correlated with delta IL-1β | 1st study to show that exercise may have greater antidepressant efficacy in inflammatory depression. Exercise did not lower cytokine levels. Lacked control group. |
| Lavebratt et al., 2017 [119] | N = 116, all MDD (follow-up data, n = 89) | Secondary analysis of RCT comparing exercise and ICBT. 12 w exercise intervention: light, moderate or vigorous | IL-6 | Higher baseline levels of IL-6 associated with greater improvement in depressive symptom severity. Positive correlation (p = 0.049) between reduced symptoms and reduction in IL-6 level | No significant effect of exercise intensities on IL-6 change. Exercise outside the supervised exercise not monitored. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suneson, K.; Lindahl, J.; Chamli Hårsmar, S.; Söderberg, G.; Lindqvist, D. Inflammatory Depression—Mechanisms and Non-Pharmacological Interventions. Int. J. Mol. Sci. 2021, 22, 1640. https://doi.org/10.3390/ijms22041640
Suneson K, Lindahl J, Chamli Hårsmar S, Söderberg G, Lindqvist D. Inflammatory Depression—Mechanisms and Non-Pharmacological Interventions. International Journal of Molecular Sciences. 2021; 22(4):1640. https://doi.org/10.3390/ijms22041640
Chicago/Turabian StyleSuneson, Klara, Jesper Lindahl, Simon Chamli Hårsmar, Gustav Söderberg, and Daniel Lindqvist. 2021. "Inflammatory Depression—Mechanisms and Non-Pharmacological Interventions" International Journal of Molecular Sciences 22, no. 4: 1640. https://doi.org/10.3390/ijms22041640
APA StyleSuneson, K., Lindahl, J., Chamli Hårsmar, S., Söderberg, G., & Lindqvist, D. (2021). Inflammatory Depression—Mechanisms and Non-Pharmacological Interventions. International Journal of Molecular Sciences, 22(4), 1640. https://doi.org/10.3390/ijms22041640