
Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Subjects
2.2. Genetic Analysis
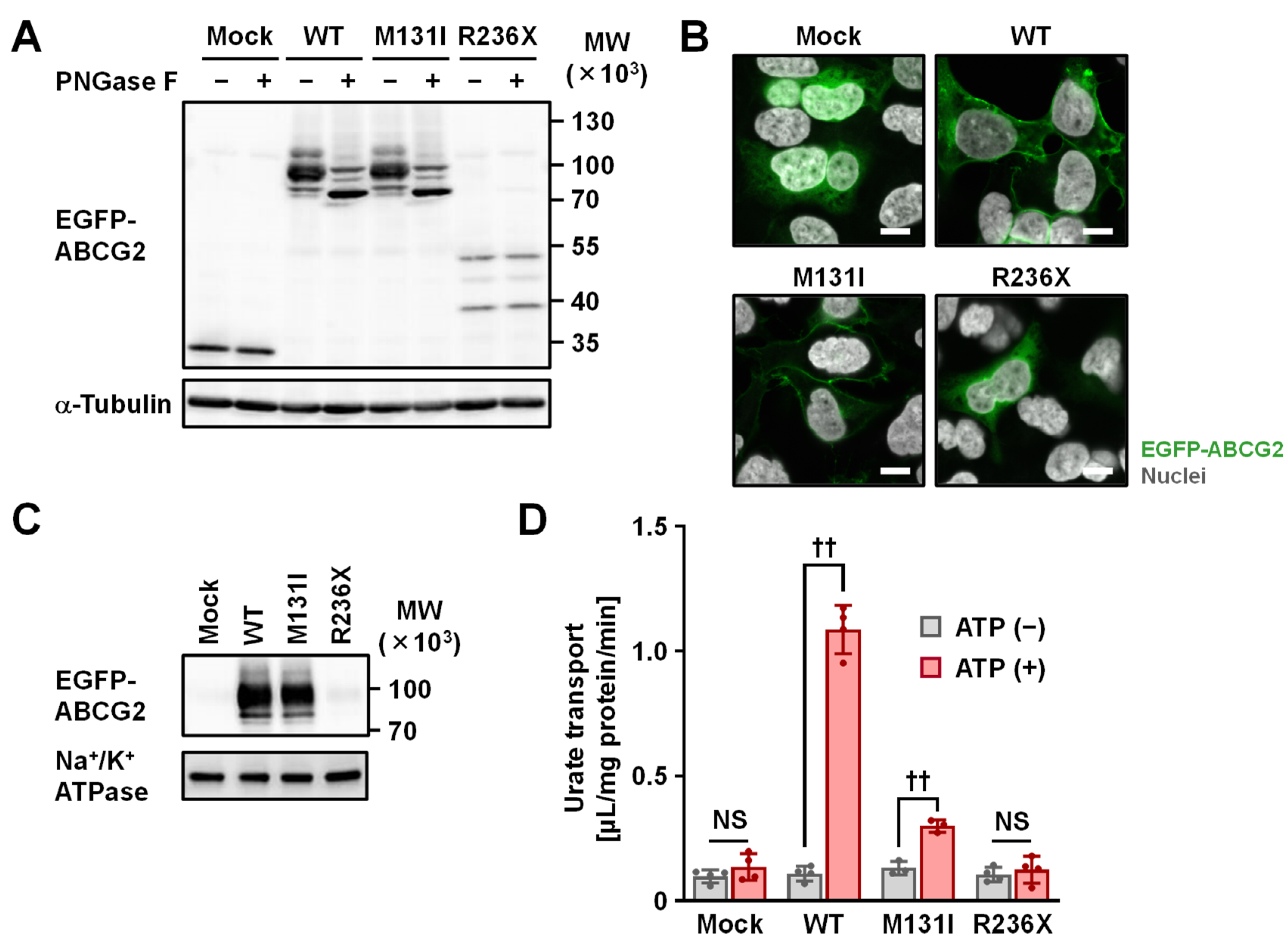
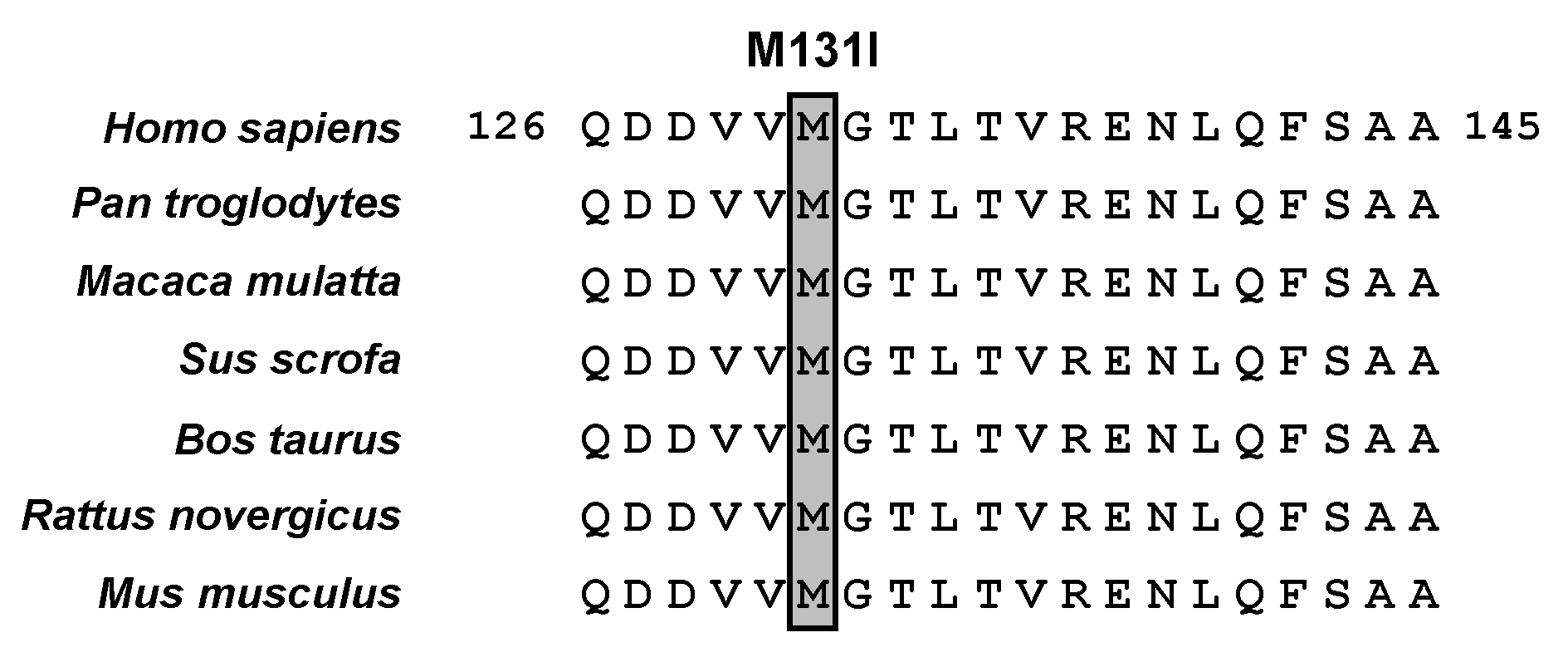
2.3. Functional Analysis
3. Discussion
4. Materials and Methods
4.1. Clinical Subjects
4.2. Clinical Investigations and Sequence Analyses
4.3. Materials
4.4. Preparation of ABCG2 Variants Expression Vector
4.5. Cell Culture and Transfection
4.6. Preparation of Whole-Cell Lysates
4.7. Preparation of ABCG2-Expressing Plasma Membrane Vesicles
4.8. Immunoblotting
4.9. Confocal Laser Scanning Microscopic Observation
4.10. Urate Transport Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG2 | ATP-binding cassette transporter G2 |
| ATP | Adenosine triphosphate |
| FE-UA | Fractional excretion of uric acid |
| GLUT9 | Glucose transporter 9 |
| MAF | Minor allele frequency |
| SUA | Serum uric acid |
| URAT1 | Urate transporter 1 |
| WT | Wild-type |
References
- Dalbeth, N.; Choi, H.K.; Joosten, L.A.B.; Khanna, P.P.; Matsuo, H.; Perez-Ruiz, F.; Stamp, L.K. Gout. Nat. Rev. Dis. Primers 2019, 5, 69. [Google Scholar] [CrossRef]
- Kawamura, Y.; Nakaoka, H.; Nakayama, A.; Okada, Y.; Yamamoto, K.; Higashino, T.; Sakiyama, M.; Shimizu, T.; Ooyama, H.; Ooyama, K.; et al. Genome-wide association study revealed novel loci which aggravate asymptomatic hyperuricaemia into gout. Ann. Rheum. Dis. 2019, 78, 1430–1437. [Google Scholar] [CrossRef]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.F.; Grainge, M.J.; See, L.C.; Yu, K.H.; Luo, S.F.; Zhang, W.; Doherty, M. Epidemiology and management of gout in Taiwan: A nationwide population study. Arthritis Res. Ther. 2015, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Pascart, T.; Norberciak, L.; Ea, H.K.; Guggenbuhl, P.; Liote, F. Patients with early-onset gout and development of earlier severe joint involvement and metabolic comorbid conditions: Results from a cross-sectional epidemiologic survey. Arthritis Care Res. 2019, 71, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fang, W.; Zeng, X.; Zhang, Y.; Ma, Y.; Sheng, F.; Zhang, X. Clinical characteristics of early- and late-onset gout: A cross-sectional observational study from a Chinese gout clinic. Medicine 2016, 95, e5425. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [Green Version]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Hoque, K.M.; Dixon, E.E.; Lewis, R.M.; Allan, J.; Gamble, G.D.; Phipps-Green, A.J.; Halperin Kuhns, V.L.; Horne, A.M.; Stamp, L.K.; Merriman, T.R.; et al. The ABCG2 Q141K hyperuricemia and gout associated variant illuminates the physiology of human urate excretion. Nat. Commun. 2020, 11, 2767. [Google Scholar] [CrossRef]
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 341–353. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Nakaoka, H.; Nakamura, T.; Nakashima, H.; Takada, Y.; Oikawa, Y.; Takada, T.; Sakiyama, M.; Shimizu, S.; et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 2014, 4, 5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaming, M.L.; Lagas, J.S.; Schinkel, A.H. Physiological and pharmacological roles of ABCG2 (BCRP): Recent findings in Abcg2 knockout mice. Adv. Drug Deliv. Rev. 2009, 61, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Hegedus, T. The ABCG2/BCRP transporter and its variants—From structure to pathology. FEBS Lett. 2020, 594, 4012–4034. [Google Scholar] [CrossRef]
- Allikmets, R.; Schriml, L.M.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar] [PubMed]
- Takada, T.; Yamamoto, T.; Matsuo, H.; Tan, J.K.; Ooyama, K.; Sakiyama, M.; Miyata, H.; Yamanashi, Y.; Toyoda, Y.; Higashino, T.; et al. Identification of ABCG2 as an exporter of uremic toxin indoxyl sulfate in mice and as a crucial factor influencing CKD progression. Sci. Rep. 2018, 8, 11147. [Google Scholar] [CrossRef]
- Heyes, N.; Kapoor, P.; Kerr, I.D. Polymorphisms of the multidrug pump ABCG2: A systematic review of their effect on protein expression, function, and drug pharmacokinetics. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1886–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiburkova, B.; Pavelcova, K.; Zavada, J.; Petru, L.; Simek, P.; Cepek, P.; Pavlikova, M.; Matsuo, H.; Merriman, T.R.; Pavelka, K. Functional non-synonymous variants of ABCG2 and gout risk. Rheumatology 2017, 56, 1982–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashino, T.; Takada, T.; Nakaoka, H.; Toyoda, Y.; Stiburkova, B.; Miyata, H.; Ikebuchi, Y.; Nakashima, H.; Shimizu, S.; Kawaguchi, M.; et al. Multiple common and rare variants of ABCG2 cause gout. RMD Open 2017, 3, e000464. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, K.; Itoda, M.; Sai, K.; Saito, Y.; Kaniwa, N.; Shirao, K.; Hamaguchi, T.; Kunitoh, H.; Yamamoto, N.; Tamura, T.; et al. Genetic variation and haplotype structure of the ABC transporter gene ABCG2 in a Japanese population. Drug Metab. Pharmacokinet. 2006, 21, 109–121. [Google Scholar] [CrossRef]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, F.; Narang, R.K.; Phipps-Green, A.; Gamble, G.G.; Tausche, A.K.; So, A.; Riches, P.; Andres, M.; Perez-Ruiz, F.; Doherty, M.; et al. Systematic genetic analysis of early-onset gout: ABCG2 is the only associated locus. Rheumatology 2020, 59, 2544–2549. [Google Scholar] [CrossRef]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Jesina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. Ther. 2019, 21, 77. [Google Scholar] [CrossRef] [Green Version]
- Hurba, O.; Mancikova, A.; Krylov, V.; Pavlikova, M.; Pavelka, K.; Stiburkova, B. Complex analysis of urate transporters SLC2A9, SLC22A12 and functional characterization of non-synonymous allelic variants of GLUT9 in the Czech population: No evidence of effect on hyperuricemia and gout. PLoS ONE 2014, 9, e107902. [Google Scholar] [CrossRef]
- Pavelcova, K.; Bohata, J.; Pavlikova, M.; Bubenikova, E.; Pavelka, K.; Stiburkova, B. Evaluation of the influence of genetic variants of SLC2A9 (GLUT9) and SLC22A12 (URAT1) on the development of hyperuricemia and gout. J. Clin. Med. 2020, 9, 2510. [Google Scholar] [CrossRef]
- Toyoda, Y.; Pavelcova, K.; Klein, M.; Suzuki, H.; Takada, T.; Stiburkova, B. Familial early-onset hyperuricemia and gout associated with a newly identified dysfunctional variant in urate transporter ABCG2. Arthritis Res. Ther. 2019, 21, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiburkova, B.; Sebesta, I.; Ichida, K.; Nakamura, M.; Hulkova, H.; Krylov, V.; Kryspinova, L.; Jahnova, H. Novel allelic variants and evidence for a prevalent mutation in URAT1 causing renal hypouricemia: Biochemical, genetics and functional analysis. Eur. J. Hum. Genet. 2013, 21, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of human ABCG2: From technical background to recent updates with clinical implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Miyata, H.; Takada, T.; Toyoda, Y.; Matsuo, H.; Ichida, K.; Suzuki, H. Identification of febuxostat as a new strong ABCG2 inhibitor: Potential applications and risks in clinical situations. Front. Pharmacol. 2016, 7, 518. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, Y.; Mancikova, A.; Krylov, V.; Morimoto, K.; Pavelcova, K.; Bohata, J.; Pavelka, K.; Pavlikova, M.; Suzuki, H.; Matsuo, H.; et al. Functional characterization of clinically-relevant rare variants in ABCG2 identified in a gout and hyperuricemia cohort. Cells 2019, 8, 363. [Google Scholar] [CrossRef] [Green Version]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Toyoda, Y.; Wakabayashi-Nakao, K.; Tamaki, H.; Osumi, M.; Ishikawa, T. Ubiquitin-mediated proteasomal degradation of ABC transporters: A new aspect of genetic polymorphisms and clinical impacts. J. Pharm. Sci. 2011, 100, 3602–3619. [Google Scholar] [CrossRef]
- Eckenstaler, R.; Benndorf, R.A. 3D structure of the transporter ABCG2-What’s new? Br. J. Pharmacol. 2020, 177, 1485–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, Y.; Takada, T.; Miyata, H.; Ishikawa, T.; Suzuki, H. Regulation of the axillary osmidrosis-associated ABCC11 protein stability by N-linked glycosylation: Effect of glucose condition. PLoS ONE 2016, 11, e0157172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morar, B.; Gresham, D.; Angelicheva, D.; Tournev, I.; Gooding, R.; Guergueltcheva, V.; Schmidt, C.; Abicht, A.; Lochmuller, H.; Tordai, A.; et al. Mutation history of the Roma/Gypsies. Am. J. Hum. Genet. 2004, 75, 596–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaydjieva, L.; Morar, B.; Chaix, R.; Tang, H. A newly discovered founder population: The Roma/Gypsies. Bioessays 2005, 27, 1084–1094. [Google Scholar] [CrossRef]
- Roberts, R.L.; Wallace, M.C.; Phipps-Green, A.J.; Topless, R.; Drake, J.M.; Tan, P.; Dalbeth, N.; Merriman, T.R.; Stamp, L.K. ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharm. J. 2017, 17, 201–203. [Google Scholar] [CrossRef]
- Wen, C.C.; Yee, S.W.; Liang, X.; Hoffmann, T.J.; Kvale, M.N.; Banda, Y.; Jorgenson, E.; Schaefer, C.; Risch, N.; Giacomini, K.M. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin. Pharmacol. Ther. 2015, 97, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Horvathova, V.; Bohata, J.; Pavlikova, M.; Pavelcova, K.; Pavelka, K.; Senolt, L.; Stiburkova, B. Interaction of the p.Q141K variant of the ABCG2 gene with clinical data and cytokine levels in primary hyperuricemia and gout. J. Clin. Med. 2019, 8, 1965. [Google Scholar] [CrossRef] [Green Version]
- Lehtisalo, M.; Keskitalo, J.E.; Tornio, A.; Lapatto-Reiniluoto, O.; Deng, F.; Jaatinen, T.; Viinamaki, J.; Neuvonen, M.; Backman, J.T.; Niemi, M. Febuxostat, but not allopurinol, markedly raises the plasma concentrations of the breast cancer resistance protein substrate rosuvastatin. Clin. Transl. Sci. 2020, 13, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.S.; Ford, I.; Nuki, G.; Hallas, J.; Hawkey, C.J.; Webster, J.; Ralston, S.H.; Walters, M.; Robertson, M.; De Caterina, R.; et al. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (FAST): A multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet 2020, 396, 1745–1757. [Google Scholar] [CrossRef]
- Stiburkova, B.; Bleyer, A.J. Changes in serum urate and urate excretion with age. Adv. Chronic Kidney Dis. 2012, 19, 372–376. [Google Scholar] [CrossRef]
- Higashino, T.; Morimoto, K.; Nakaoka, H.; Toyoda, Y.; Kawamura, Y.; Shimizu, S.; Nakamura, T.; Hosomichi, K.; Nakayama, A.; Ooyama, K.; et al. Dysfunctional missense variant of OAT10/SLC22A13 decreases gout risk and serum uric acid levels. Ann. Rheum. Dis. 2020, 79, 164–166. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.L.; Robinson, H.; Masi, A.T.; Decker, J.L.; McCarty, D.J.; Yu, T.F. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977, 20, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Mraz, M.; Hurba, O.; Bartl, J.; Dolezel, Z.; Marinaki, A.; Fairbanks, L.; Stiburkova, B. Modern diagnostic approach to hereditary xanthinuria. Urolithiasis 2015, 43, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Ichida, K.; Sebesta, I. Novel homozygous insertion in SLC2A9 gene caused renal hypouricemia. Mol. Genet. Metab. 2011, 102, 430–435. [Google Scholar] [CrossRef]
- Stiburkova, B.; Gabrikova, D.; Cepek, P.; Simek, P.; Kristian, P.; Cordoba-Lanus, E.; Claverie-Martin, F. Prevalence of URAT1 allelic variants in the Roma population. Nucleosides Nucleotides Nucleic Acids 2016, 35, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Khunweeraphong, N.; Szollosi, D.; Stockner, T.; Kuchler, K. The ABCG2 multidrug transporter is a pump gated by a valve and an extracellular lid. Nat. Commun. 2019, 10, 5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orban, T.I.; Seres, L.; Ozvegy-Laczka, C.; Elkind, N.B.; Sarkadi, B.; Homolya, L. Combined localization and real-time functional studies using a GFP-tagged ABCG2 multidrug transporter. Biochem. Biophys. Res. Commun. 2008, 367, 667–673. [Google Scholar] [CrossRef]
- Takada, T.; Suzuki, H.; Sugiyama, Y. Characterization of polarized expression of point- or deletion-mutated human BCRP/ABCG2 in LLC-PK1 cells. Pharm. Res. 2005, 22, 458–464. [Google Scholar] [CrossRef]
- Stiburkova, B.; Miyata, H.; Zavada, J.; Tomcik, M.; Pavelka, K.; Storkanova, G.; Toyoda, Y.; Takada, T.; Suzuki, H. Novel dysfunctional variant in ABCG2 as a cause of severe tophaceous gout: Biochemical, molecular genetics and functional analysis. Rheumatology 2016, 55, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, Y.; Sakurai, A.; Mitani, Y.; Nakashima, M.; Yoshiura, K.; Nakagawa, H.; Sakai, Y.; Ota, I.; Lezhava, A.; Hayashizaki, Y.; et al. Earwax, osmidrosis, and breast cancer: Why does one SNP (538G>A) in the human ABC transporter ABCC11 gene determine earwax type? FASEB J. 2009, 23, 2001–2013. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wakabayashi-Nakao, K.; Tamura, A.; Toyoda, Y.; Koshiba, S.; Ishikawa, T. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 2009, 276, 7237–7252. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Proband | Mother | Maternal uncle | Maternal grandfather | Father | |
|---|---|---|---|---|---|
| Age of onset (HA/gout) | 12 years (HA) | 35 years (HA) | 35 years (gout) | 43 years (gout) | 30–35 (HA) |
| Main symptoms | Asymptomatic | Asymptomatic | Gout | Gout | Asymptomatic |
| Other symptoms or diseases | Astigmatism; Bilateral cataracts; Myopia; Bronchial asthma; Obesity | Thyropathy; Vertebrogenic Algic syndrome | T2D; Hypertension; Obesity; Thyropathy | T2D; Hypertension | Bronchial asthma; Hypertriglyceridemia; Hypertension; Central obesity |
| SUA before treatment [µmol/L (mg/dL)] | 397–405 (6.67–6.81) | 420–439 (7.06–7.38) | >500 (>8.41) | 537 (9.03) | 487 (8.19) |
| UUA [mmol/mol creatinine] | 3.34 | 1.47 | 2.10 | n.d. | 2.45 |
| FE-UA [%] | 2.2 | 4.6 | 2.9 | n.d. | 3.9 |
| Therapy | No | No | Allopurinol (100 mg per day) | Colchicine; Allopurinol (300 mg per day) | No |
| SUA during treatment [µmol/L (mg/dL)] | n.d. | n.d. | 446 (7.50) (non-compliance) | 422 (7.09) (non-compliance) | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toyoda, Y.; Pavelcová, K.; Bohatá, J.; Ješina, P.; Kubota, Y.; Suzuki, H.; Takada, T.; Stiburkova, B. Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout. Int. J. Mol. Sci. 2021, 22, 1935. https://doi.org/10.3390/ijms22041935
Toyoda Y, Pavelcová K, Bohatá J, Ješina P, Kubota Y, Suzuki H, Takada T, Stiburkova B. Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout. International Journal of Molecular Sciences. 2021; 22(4):1935. https://doi.org/10.3390/ijms22041935
Chicago/Turabian StyleToyoda, Yu, Kateřina Pavelcová, Jana Bohatá, Pavel Ješina, Yu Kubota, Hiroshi Suzuki, Tappei Takada, and Blanka Stiburkova. 2021. "Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout" International Journal of Molecular Sciences 22, no. 4: 1935. https://doi.org/10.3390/ijms22041935
APA StyleToyoda, Y., Pavelcová, K., Bohatá, J., Ješina, P., Kubota, Y., Suzuki, H., Takada, T., & Stiburkova, B. (2021). Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout. International Journal of Molecular Sciences, 22(4), 1935. https://doi.org/10.3390/ijms22041935