
Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice


 , , , ,
, , , ,
Abstract
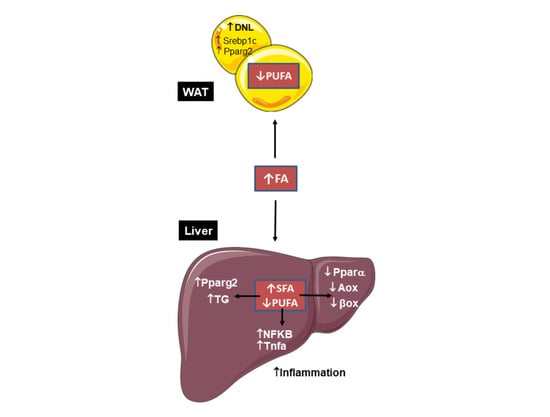
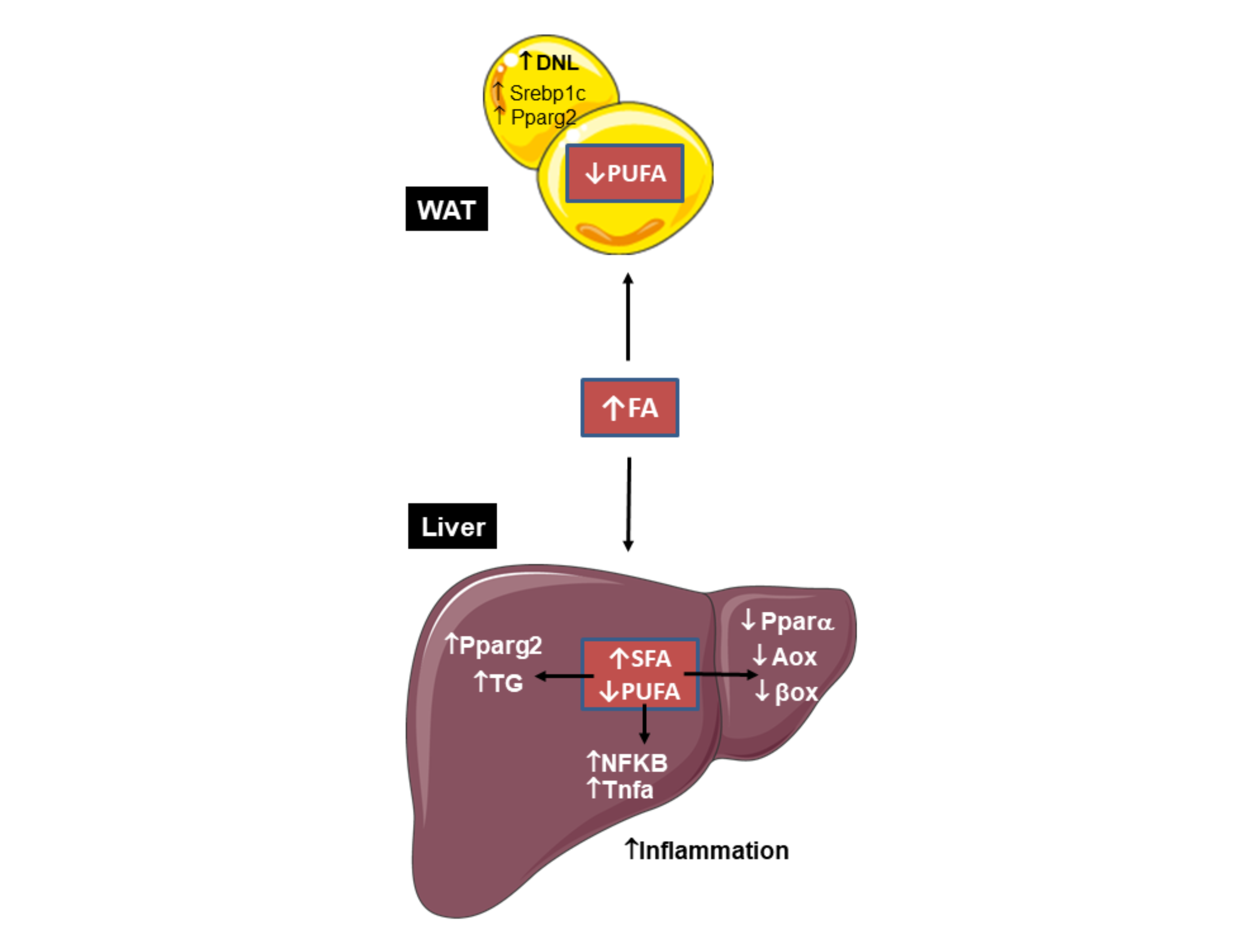
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
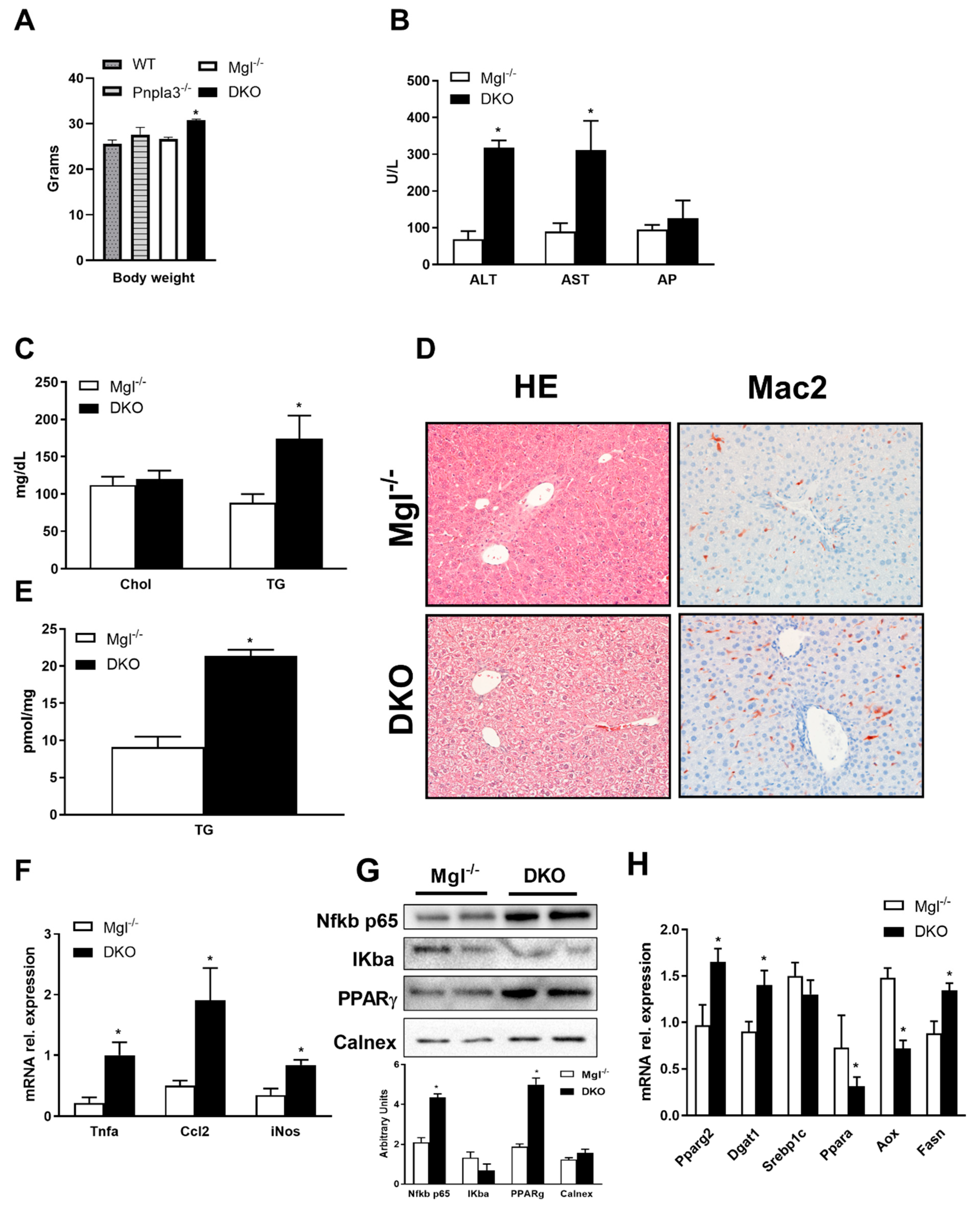
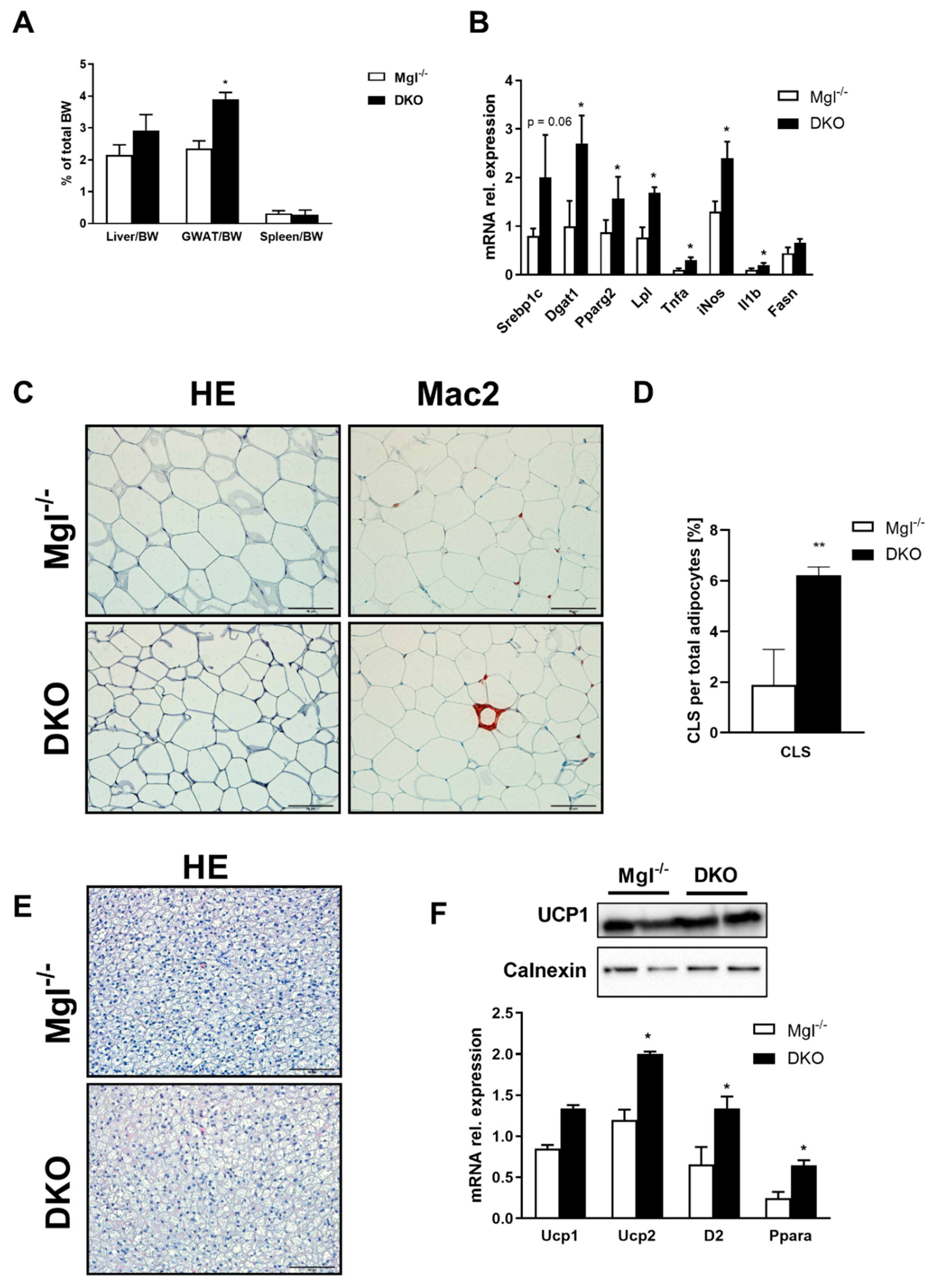
2.1. Concomitant Absence of Mgl and Pnpla3 Increases Susceptibility to Weight Gain and Hepatic Inflammation
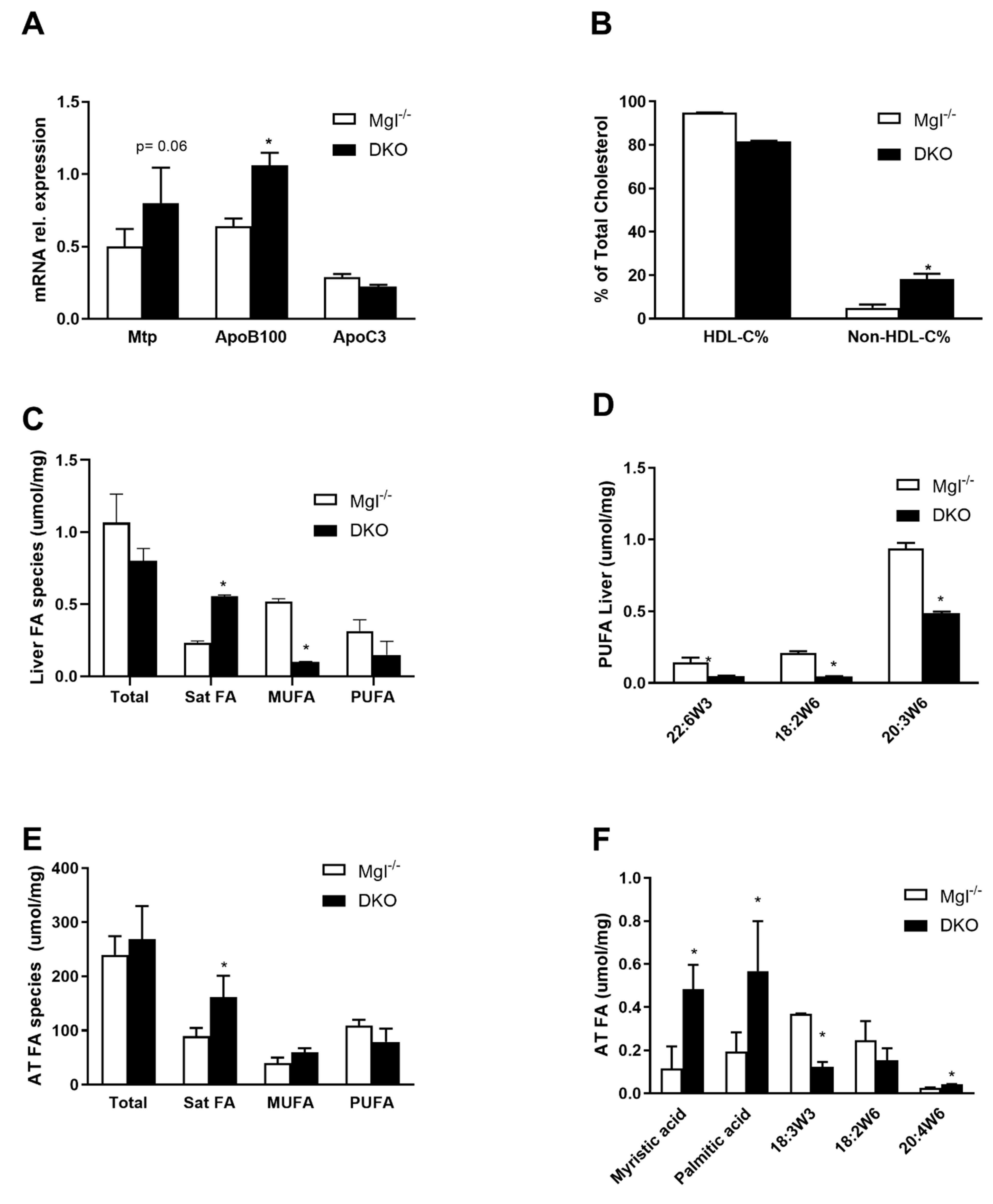
2.2. DKO Mice Show a Depleted Pool of Unsaturated FAs in the Liver with Enriched Saturated Species in the Liver and AT
2.3. Absence of Mgl and Pnpla3 Increases White Adipose Tissue Lipid Storage and Inflammation
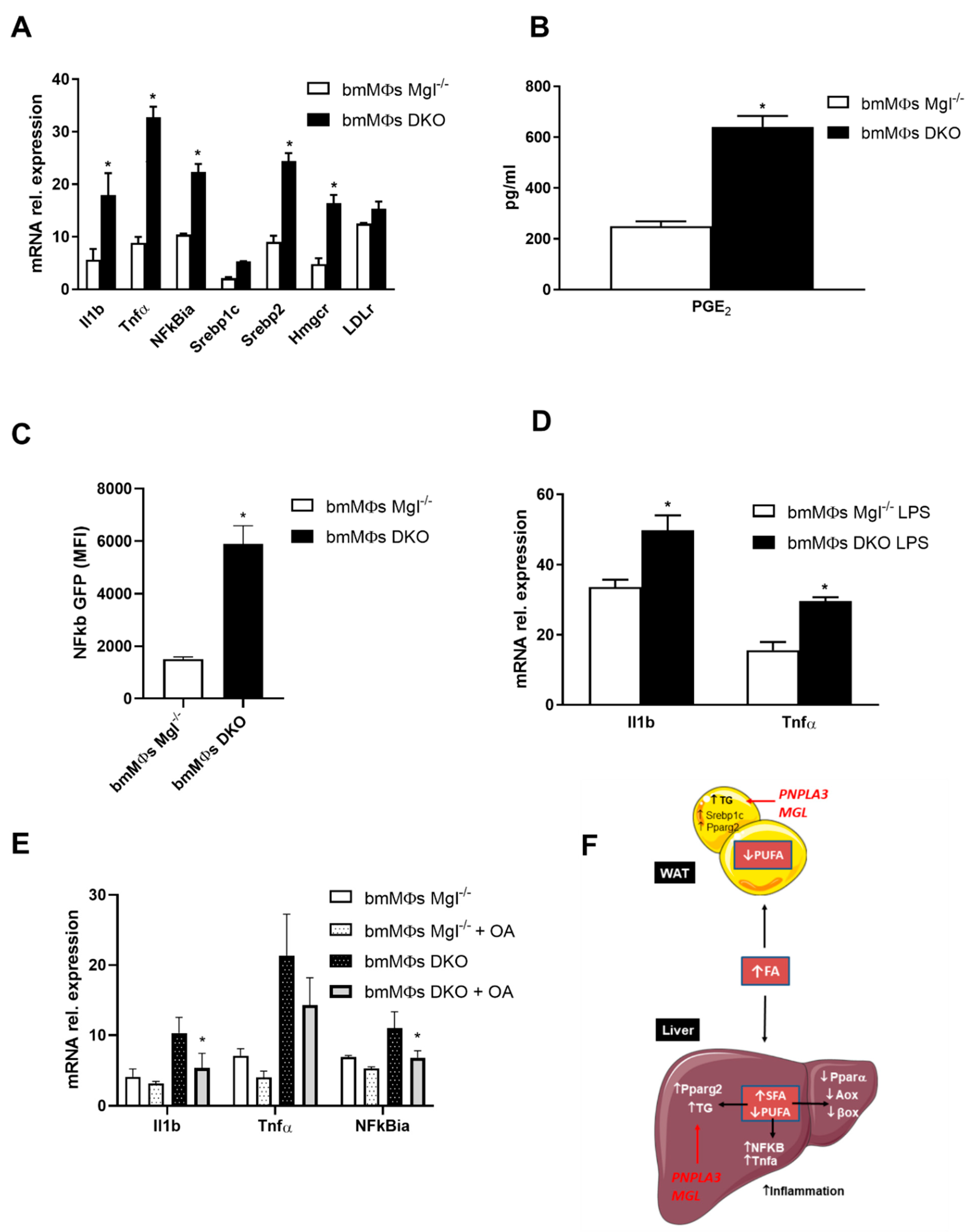
2.4. Primary BMDMs from DKO Mice Are Proinflammatory and Their Phenotypes Can Be Partially Reversed with Oleic Acid Treatment
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Serum Biochemistry
4.3. Liver Histology, Immunohistochemistry, and CLS Quantification
4.4. Western Blotting
4.5. Gas Chromatography
4.6. Gene Expression
4.7. Hepatic Lipid Concentrations
4.8. BMDMs Isolation LPS and OA Treatments
4.9. U937-NF-κB GFP
4.10. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MGL | Monoacylglycerol lipase; |
| PNPLA3 | the patatin-like phospholipase domain containing 3; |
| HFD | High-fat diet; |
| TG | Triglyceride; |
| FA | Fatty acid; |
| AA | Arachidonic acid; |
| NR | Nuclear receptor; |
| WT | Wild type; |
| Mgl−/− | Mgl knockout; |
| MG | Monoacylglycerol; |
| Ppara | Peroxisome proliferator activated receptor alpha; |
| Pparg | Peroxisome proliferator activated receptor gamma; |
| Ldlr | LDL receptor; |
| Hmgcr | 3-hydroxy-3-methyl-glutaryl-coA reductase; |
| Srebp1c | Sterol regulatory element-binding protein 1c; |
| Srebp2 | Sterol regulatory element-binding protein 2; |
| Fasn | Fatty acid synthase; |
| Ccl2 | C-C Motif Chemokine Ligand 2; |
| Tnfa | Tumor necrosis factor alpha; |
| MTP | Microsomal triglyceride transfer protein; |
| Apob100 | Apolipoprotein b100; |
| Apoc3 | Apolipoprotein c3; |
| Il1b | Interleukin 1 beta; |
| Ucp1/2 | Uncoupling Protein 1/2; |
| D2 | Dopamine receptor 2; |
| ALT | Alanine aminotransferase; |
| AST | Aspartate aminotransferase; |
| SFA | Saturated fatty acids; |
| MUFA | Monounsaturated fatty acids; |
| PUFA | Polyunsaturated fatty acids; |
| CLS | Crown-like structure; |
| BAT | Brown adipose tissue; |
| PGE2 | prostaglandin E2; |
| BA | Bile acid. |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 10, 686–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Papatheodoridis, G.V.; Archimandritis, A.J. Adipokines in nonalcoholic steatohepatitis: From pathogenesis to implications in diagnosis and therapy. Mediat. Inflamm. 2009, 2009, 831670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Claudel, T.; Trauner, M. PNPLA3 expression and its impact on the liver: Current perspectives. Hepatic Med. Evid. Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, P.; Romeo, S. The role of PNPLA3 in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 900–906. [Google Scholar] [CrossRef]
- Tardelli, M.; Bruschi, F.V.; Trauner, M. The role of metabolic lipases in the pathogenesis and management of liver disease. Hepatology 2020, 72, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Starlinger, P.; Marra, F.; Trauner, M. PNPLA3 I148M variant impairs liver X receptor signaling and cholesterol homeostasis in human hepatic stellate cells. Hepatol. Commun. 2019, 3, 1191–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018, 68, 522–532. [Google Scholar] [CrossRef]
- Tardelli, M.; Bruschi, F.V.; Fuchs, C.D.; Claudel, T.; Auer, N.; Kunczer, V.; Baumgartner, M.; Ronda, O.A.; Verkade, H.J.; Stojakovic, T.; et al. Monoacylglycerol lipase inhibition protects from liver injury in mouse models of sclerosing cholangitis. Hepatology 2019, 71, 1750–1765. [Google Scholar] [CrossRef]
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.D.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.F.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292. [Google Scholar] [CrossRef]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.-P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, Y.; Patsenker, E.; Fickert, P.; Trauner, M.; Schuppan, D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J. Hepatol. 2005, 43, 1045–1054. [Google Scholar] [CrossRef]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; De Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.C.; Sun, Y.; Li, J.-L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Yang, X.; Liu, J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle 2010, 9, 2719–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Mottillo, E.P.; Mladenovic-Lucas, L.; Zhou, L.; Granneman, J.G. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat. Metab. 2019, 1, 560–569. [Google Scholar] [CrossRef]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Tardelli, M. Monoacylglycerol lipase reprograms lipid precursors signaling in liver disease. World J. Gastroenterol. 2020, 26, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; Basuray, S.; Li, J.; Huang, Y.; Lai, K.M.V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.-C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI 2019, 4, e127902. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. JCI 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zhang, X.; Lau, J.K.-C.; Fu, K.; Lau, H.C.H.; Xu, W.; Chu, E.S.; Lan, H.-Y.; Yu, J. Bone marrow-derived macrophage contributes to fibrosingsteatohepatitis through activating hepatic stellate cells. J. Pathol. 2019, 488, 488–500. [Google Scholar] [CrossRef] [Green Version]
- Tardelli, M.; Zeyda, K.; Moreno-Viedma, V.; Wanko, B.; Grün, N.G.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol. Metab. 2016, 5, 1131–1137. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tardelli, M.; Bruschi, F.V.; Fuchs, C.D.; Claudel, T.; Auer, N.; Kunczer, V.; Ronda, O.A.H.O.; Verkade, H.J.; Stojakovic, T.; Scharnagl, H.; et al. Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice. Int. J. Mol. Sci. 2021, 22, 2126. https://doi.org/10.3390/ijms22042126
Tardelli M, Bruschi FV, Fuchs CD, Claudel T, Auer N, Kunczer V, Ronda OAHO, Verkade HJ, Stojakovic T, Scharnagl H, et al. Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice. International Journal of Molecular Sciences. 2021; 22(4):2126. https://doi.org/10.3390/ijms22042126
Chicago/Turabian StyleTardelli, Matteo, Francesca V. Bruschi, Claudia D. Fuchs, Thierry Claudel, Nicole Auer, Victoria Kunczer, Onne A. H. O. Ronda, Henkjan J. Verkade, Tatjana Stojakovic, Hubert Scharnagl, and et al. 2021. "Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice" International Journal of Molecular Sciences 22, no. 4: 2126. https://doi.org/10.3390/ijms22042126
APA StyleTardelli, M., Bruschi, F. V., Fuchs, C. D., Claudel, T., Auer, N., Kunczer, V., Ronda, O. A. H. O., Verkade, H. J., Stojakovic, T., Scharnagl, H., & Trauner, M. (2021). Absence of Adiponutrin (PNPLA3) and Monoacylglycerol Lipase Synergistically Increases Weight Gain and Aggravates Steatohepatitis in Mice. International Journal of Molecular Sciences, 22(4), 2126. https://doi.org/10.3390/ijms22042126