
Extensive Placental Methylation Profiling in Normal Pregnancies

 , , , , , , , , ,
, , , , , , , , , 
 and
and
Abstract
:1. Introduction
2. Results
2.1. Study Design
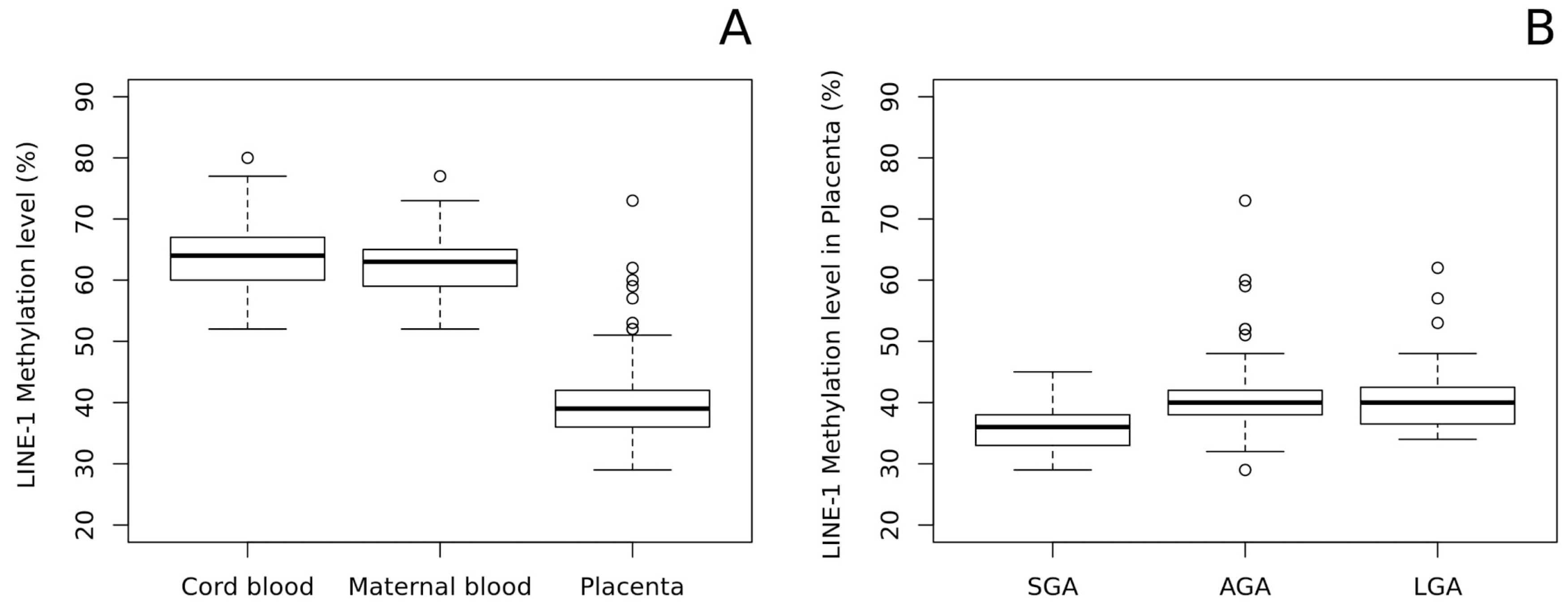
2.2. LINE-1 Methylation Analysis
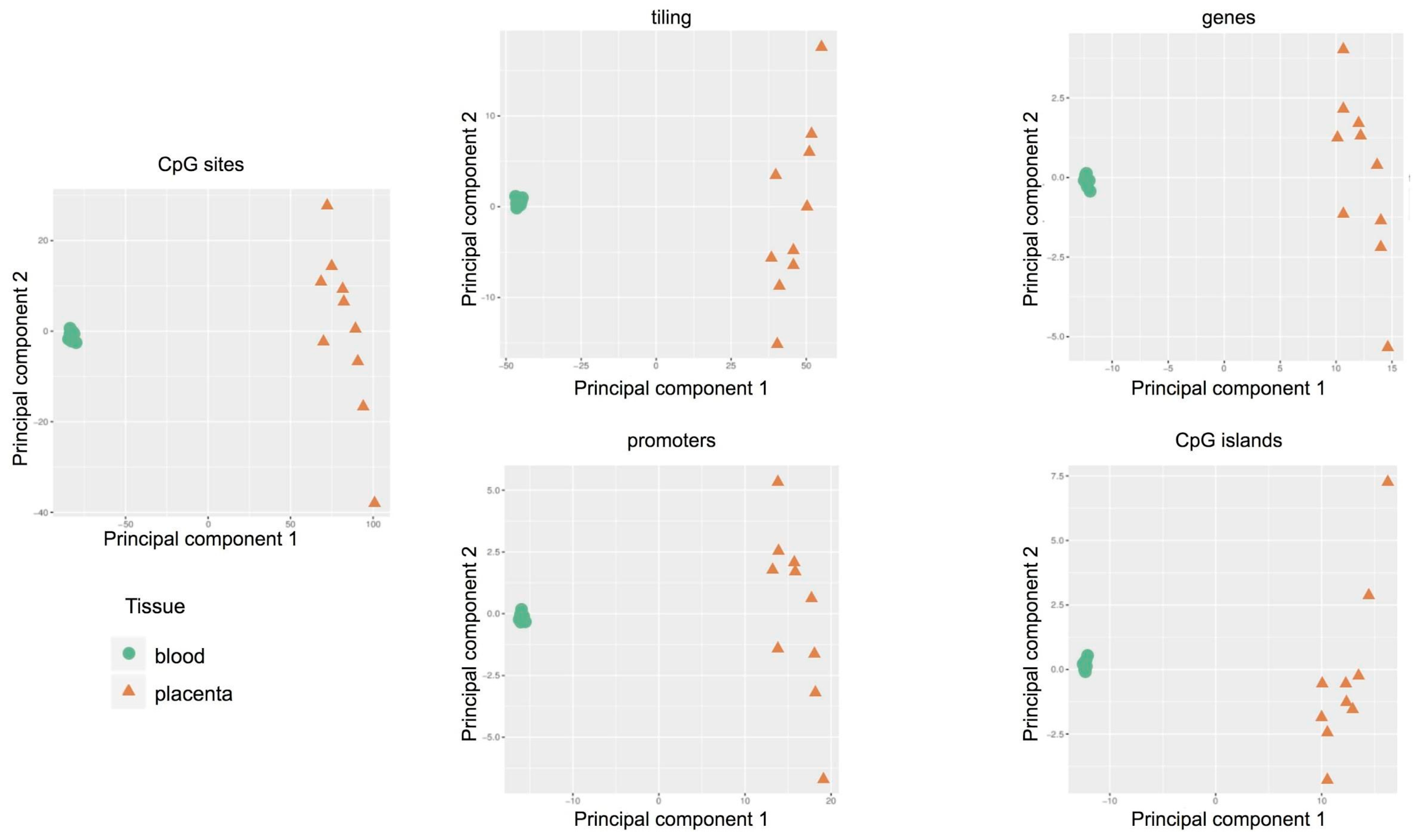
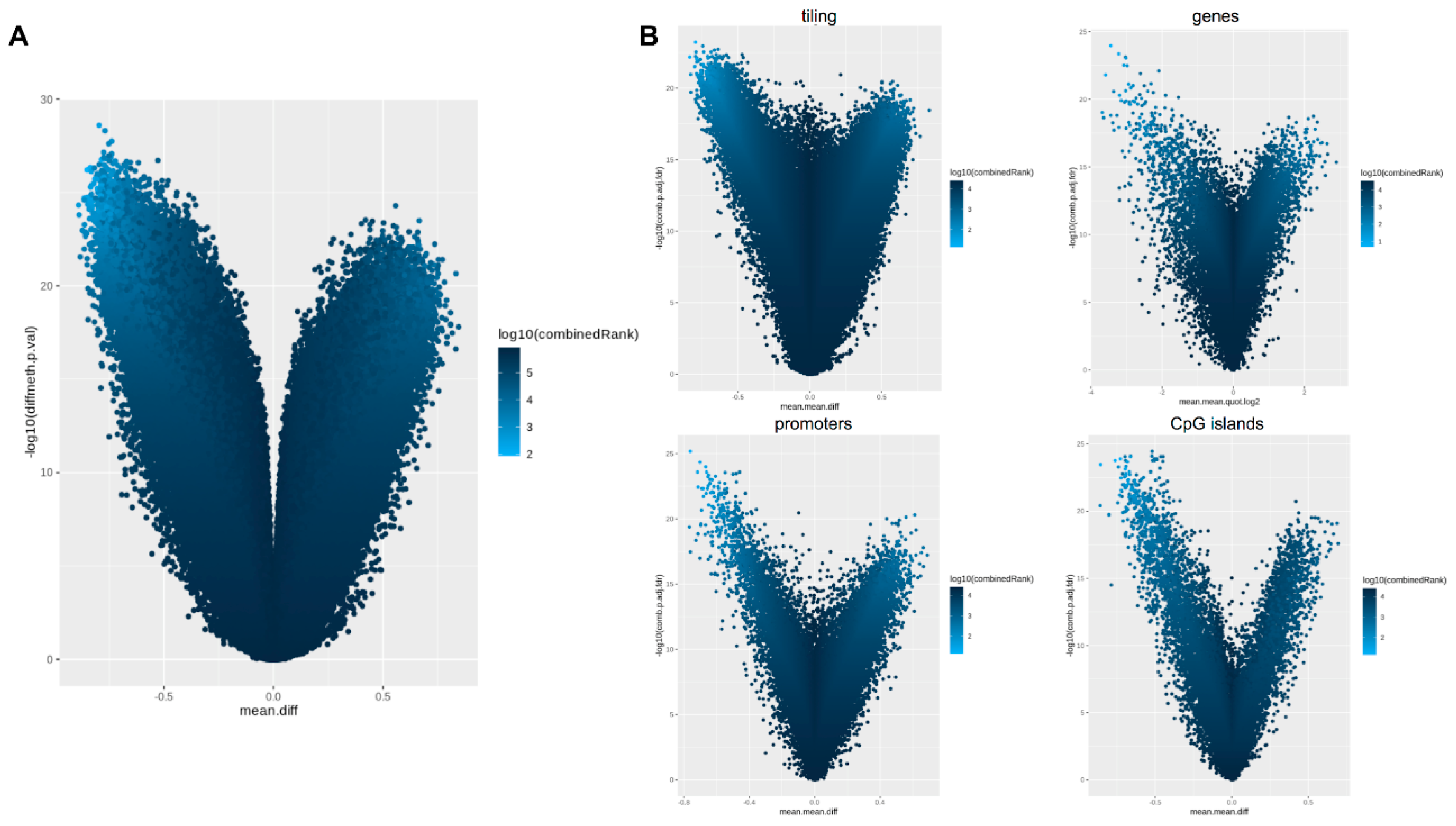
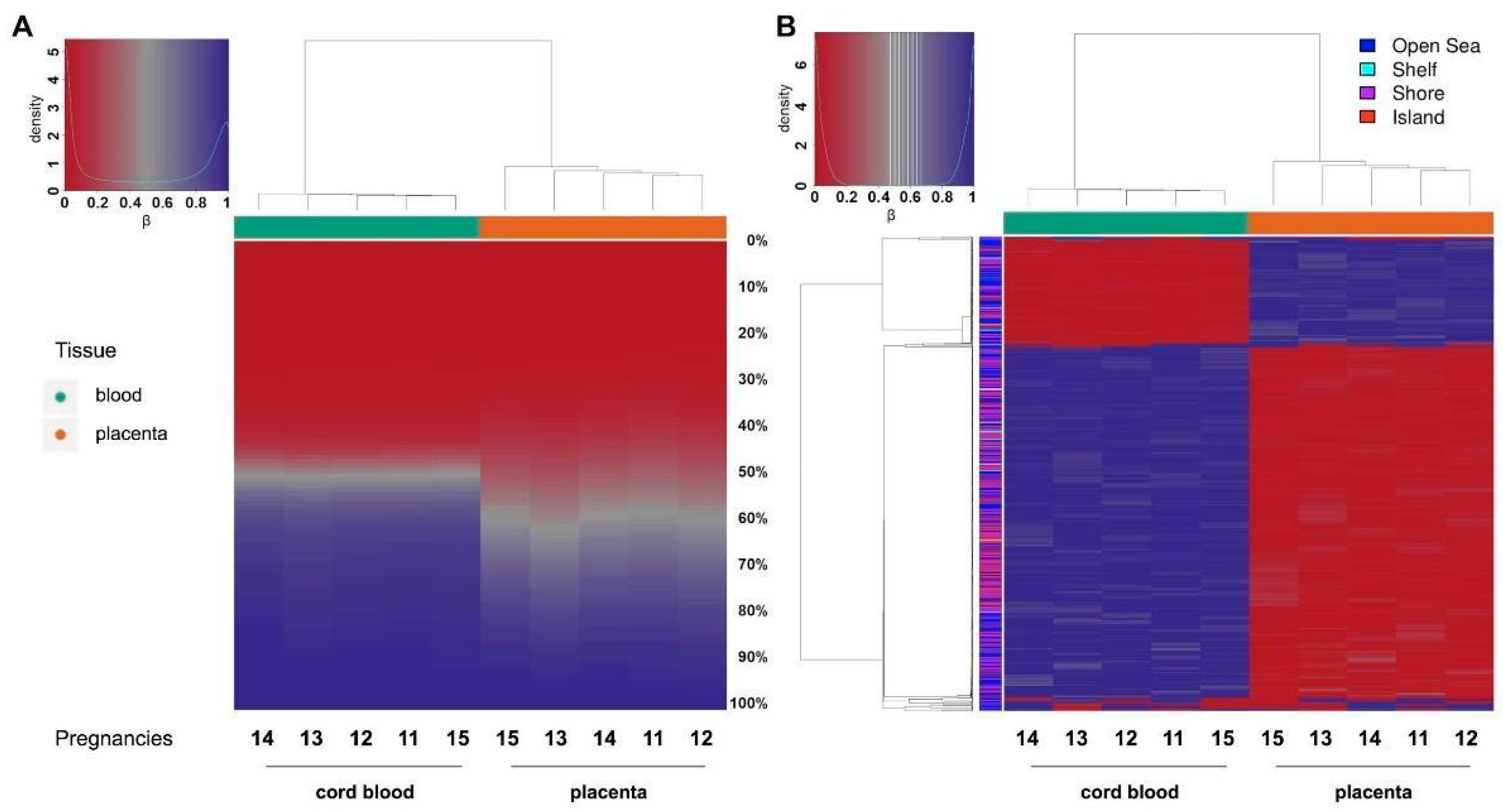
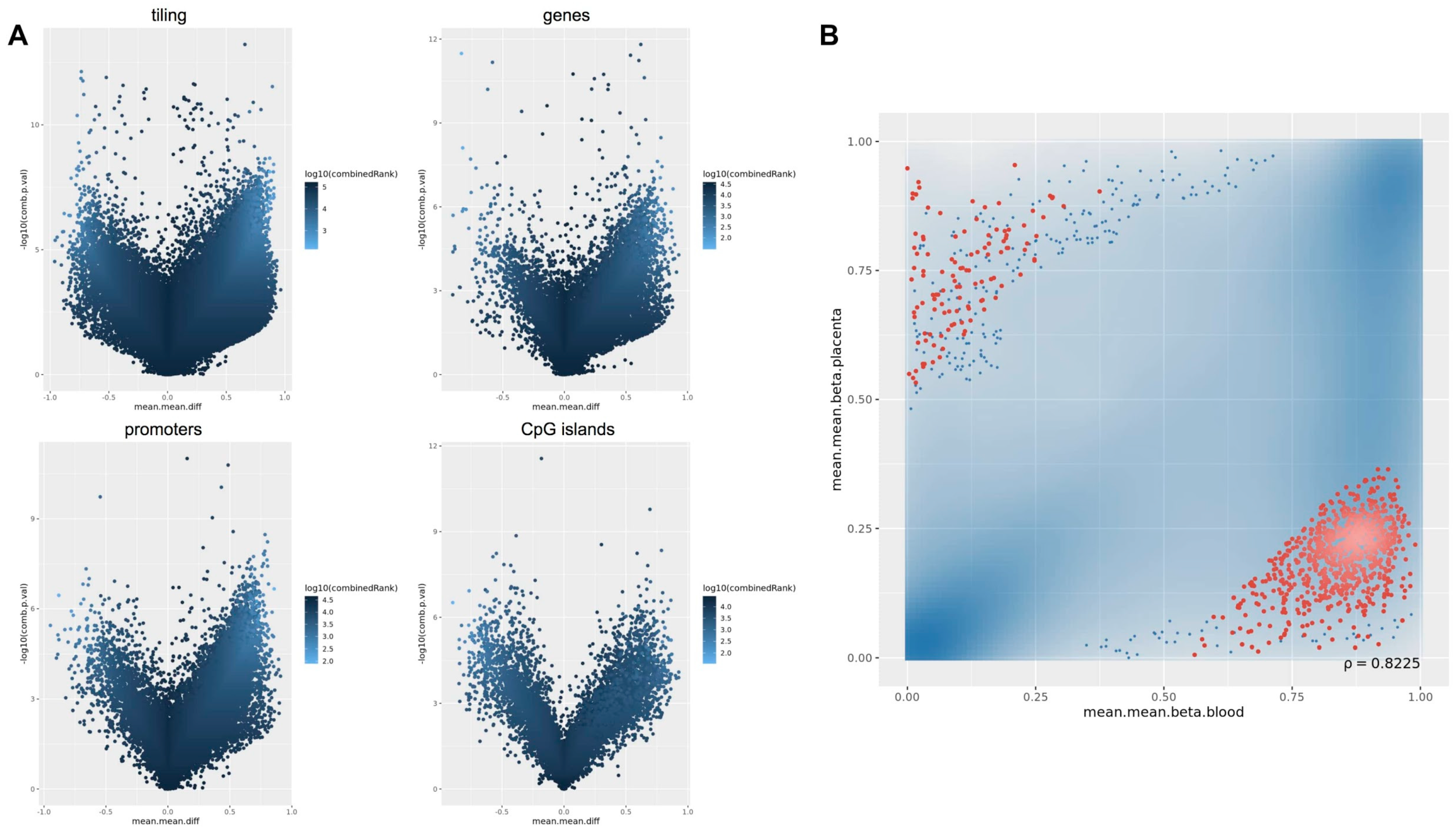
2.3. Methylation-Profiling Array
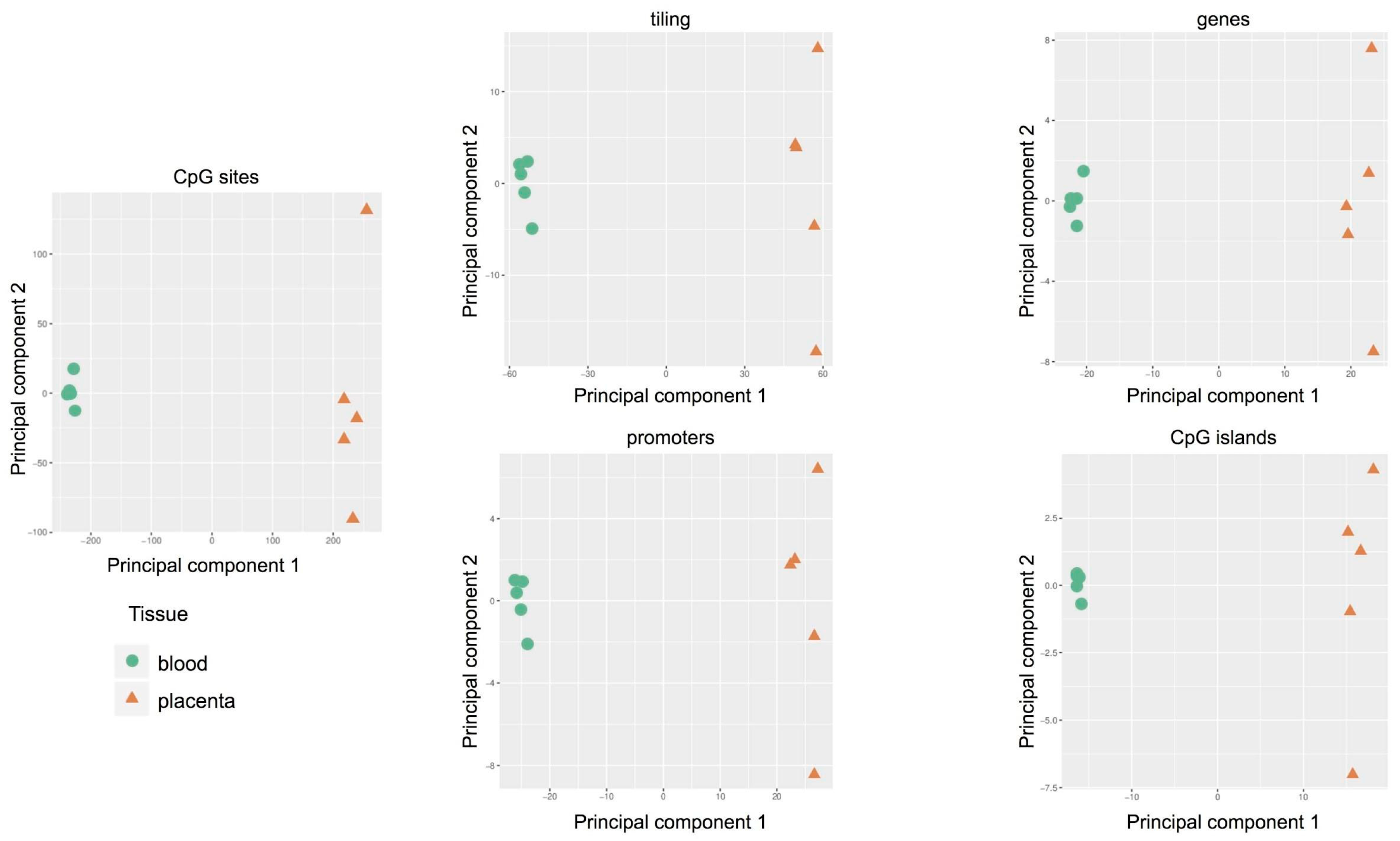
2.4. Targeted Methylation Sequencing by NGS
2.5. Comparison of the Methylation Profiles Obtained by Microarray and Targeted Sequencing
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Sample Collection and DNA Extraction
4.3. Methylation Profiling
4.3.1. LINE-1 Methylation Analysis
4.3.2. Methylation-Profiling Array
Genome-Wide Methylation Analysis
Data Management, Pre-Processing, Normalization, and Quality Control
4.3.3. Targeted Methylation Sequencing
Methylation-Capture Sequencing
Data Management, Pre-Preprocessing, Normalisation and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.D.; Godfrey, K.M.; Kirkwood, T.B.L.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef]
- Turco, M.Y.; Moffett, A. Development of the human placenta. Development 2019, 146, dev163428. [Google Scholar] [CrossRef] [Green Version]
- Miozzo, M.; Grati, F.; Bulfamante, G.; Rossella, F.; Cribiù, M.; Radaelli, T.; Cassani, B.; Persico, T.; Cetin, I.; Pardi, G.; et al. Post-zygotic origin of complete maternal chromosome 7 Isodisomy and consequent loss of placental PEG1/MEST expression. Placenta 2001, 22, 813–821. [Google Scholar] [CrossRef]
- Grati, F.R. Chromosomal mosaicism in human feto-placental development: Implications for prenatal diagnosis. J. Clin. Med. 2014, 3, 809–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miozzo, M.; Simoni, G. The role of imprinted genes in fetal growth. Biol. Neonate 2002, 81, 217–228. [Google Scholar] [CrossRef]
- Perez-Garcia, V.; Fineberg, E.; Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J.; et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Barker, D. The Developmental origins of adult disease. J. Am. Coll. Nutr. 2004, 23, 588S–595S. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahnama, F.; Shafiei, F.; Gluckman, P.D.; Mitchell, M.D.; Lobie, P.E. Epigenetic Regulation of human trophoblastic cell migration and invasion. Endocrinology 2006, 147, 5275–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serman, L.; Vlahović, M.; Sijan, M.; Bulić-Jakus, F.; Serman, A.; Sincić, N.; Matijević, R.; Jurić-Lekić, G.; Katusić, A. The impact of 5-azacytidine on placental weight, glycoprotein pattern and pro-liferating cell nuclear antigen expression in rat placenta. Placenta 2007, 28, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Milano-Foster, J.; Ray, S.; Home, P.; Ganguly, A.; Bhattacharya, B.; Bajpai, S.; Pal, A.; Mason, C.W.; Paul, S. Regulation of human trophoblast syncytialization by histone demethylase LSD1. J. Biol. Chem. 2019, 294, 17301–17313. [Google Scholar] [CrossRef]
- Robinson, W.P.; Price, E.M. The human placental methylome. Cold Spring Harb. Perspect. Med. 2015, 5, a023044. [Google Scholar] [CrossRef]
- Novakovic, B.; Wong, N.C.; Sibson, M.; Ng, H.-K.; Morley, R.; Manuelpillai, U.; Down, T.; Rakyan, V.K.; Beck, S.; Hiendleder, S.; et al. DNA Methylation-mediated Down-regulation of DNA Methyltransferase-1 (DNMT1) is coincident with, but not essential for, global hypomethylation in human placenta. J. Biol. Chem. 2010, 285, 9583–9593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.; Hong, D.; Crary, F.; Ashwood, P.; Walker, C.; Korf, I.; Robinson, W.P.; et al. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef] [Green Version]
- Reiss, D.; Zhang, Y.; Mager, D.L. Widely variable endogenous retroviral methylation levels in human placenta. Nucleic Acids Res. 2007, 35, 4743–4754. [Google Scholar] [CrossRef]
- Price, E.M.; Cotton, A.M.; Peñaherrera, M.S.; McFadden, D.E.; Kobor, M.S.; Robinson, W.P. Different measures of “genome-wide” DNA methylation exhibit unique properties in placental and somatic tissues. Epigenetics 2012, 7, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Macaulay, E.C.; Rodger, E.J.; Stockwell, P.A.; Parry, M.F.; Roberts, H.E.; Slatter, T.L.; Hung, N.A.; Devenish, C.J.; Morison, I.M. Placental hypomethylation is more pronounced in genomic loci devoid of retroelements. G3 Genes Genomes Genet. 2016, 6, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Novakovic, B.; Yuen, R.K.; Gordon, L.; Penaherrera, M.S.; Sharkey, A.; Moffett, A.; Craig, J.M.; Robinson, W.P.; Saffery, R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genom. 2011, 12, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Gobbo, G.F.; Konwar, C.; Robinson, W.P. The significance of the placental genome and methylome in fetal and maternal health. Qual. Life Res. 2019, 139, 1183–1196. [Google Scholar] [CrossRef]
- Barker, D.; Thornburg, K. Placental programming of chronic diseases, cancer and lifespan: A review. Placenta 2013, 34, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Simpkin, A.J.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Tilling, K.; Smith, G.D.; Relton, C.L. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 2015, 24, 3752–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, G.; Hajj, H.; Rifas-Shiman, S.L.; Just, A.C.; Hivert, M.F.; Burris, H.H.; Lin, X.; Litonjua, A.A.; Oken, E.; DeMeo, D.L.; et al. Birth weight-for-gestational age is associated with DNA methylation at birth and in childhood. Clin. Epigenetics 2016, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwi Putra, S.E.; Reichetzeder, C.; Hasan, A.A.; Slowinski, T.; Chu, C.; Krämer, B.K.; Kleuser, B.; Hocher, B. Being born large for gestational age is associated with increased global placental DNA methylation. Sci. Rep. 2020, 10, 927. [Google Scholar] [CrossRef]
- Filiberto, A.C.; Maccani, M.A.; Koestler, D.C.; Wilhelm-Benartzi, C.; Avissar-Whiting, M.; Banister, C.E.; Gagne, L.A.; Marsit, C.J. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics 2011, 6, 566–572. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.C.; Choufani, S.; Grafodatskaya, D.; Butcher, D.T.; Zhao, C.; Chitayat, D.; Shuman, C.; Kingdom, J.; Keating, S.; Weksberg, R. WNT2promoter methylation in human placenta is associated with low birthweight percentile in the neonate. Epigenetics 2011, 6, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, N.; Ghalwash, M.F.; Katari, S.; Coutifaris, C.; Obradovic, Z.; Sapienza, C. DNA methylation differences at growth related genes correlate with birth weight: A molecular signature linked to developmental origins of adult disease? BMC Med. Genom. 2012, 5, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Leeuwerke, M.; Eilander, M.S.; Pruis, M.G.; Lendvai, Á.; Erwich, J.J.; Scherjon, S.A.; Plösch, T.; Eijsink, J.J. DNA methylation and expression patterns of selected genes in first-trimester placental tissue from pregnancies with small-for-gestational-age infants at birth. Biol. Reprod. 2016, 94, 37. [Google Scholar] [CrossRef] [Green Version]
- Banister, C.E.; Koestler, D.C.; Maccani, M.A.; Padbury, J.F.; Houseman, E.A.; Marsit, C.J. Infant growth restriction is associated with distinct patterns of DNA methylation in human placentas. Epigenetics 2011, 6, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.; Chu, A.; Liao, W.W.; Rubbi, L.; Janzen, C.; Hsu, F.M.; Thamotharan, S.; Ganguly, A.; Lam, L.; Montoya, D.; et al. Prenatal growth patterns and birthweight are associated with differential DNA Methylation and gene expression of cardiometabolic risk genes in human placentas: A discovery-based approach. Reprod. Sci. 2018, 25, 523–539. [Google Scholar] [CrossRef]
- Bourque, D.K.; Avila, L.; Peñaherrera, M.; von Dadelszen, P.; Robinson, W.P. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010, 31, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.B.; Harris, H.R.; Barault, L. Birthweight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS ONE 2011, 6, e25254. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.L.; Clifton, V.L.; Prentis, P.; Ewing, A.; Miller, Y.D.; Pelzer, E.S. Modulation of placental gene expression in small-for-gestational-age infants. Genes 2020, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- Tekola-Ayele, F.; Zeng, X.; Ouidir, M.; Workalemahu, T.; Zhang, C.; Delahaye, F.; Wapner, R. DNA methylation loci in placenta associated with birthweight and expression of genes relevant for early development and adult diseases. Clin. Epigenetics 2020, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Adkins, R.M.; Tylavsky, F.A.; Krushkal, J. Newborn umbilical cord blood dna methylation and gene expression levels exhibit limited association with birth weight. Chem. Biodivers. 2012, 9, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.M.; Joubert, B.R.; Wu, M.C.; Olshan, A.F.; Håberg, S.E.; Ueland, P.M.; Nystad, W.; Nilsen, R.M.; Vollset, S.E.; Peddada, S.D.; et al. Neonatal genome-wide methylation patterns in relation to birth weight in the norwegian mother and child cohort. Am. J. Epidemiol. 2014, 179, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Küpers, L.K.; Monnereau, C.; Sharp, G.C.; Yousefi, P.; Salas, L.A.; Ghantous, A.; Page, C.M.; Reese, S.E.; Wilcox, A.J.; Czamara, D.; et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birth weight. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal anthropometric charts: The italian neonatal study compared with other european studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Lin, C.W.; Park, Y.; Tseng, G. MethylSeqDesign: A framework for Methyl-Seq genome-wide power calculation and study design issues. Biostatistics 2021, 22, 35–50. [Google Scholar] [CrossRef]
- Tabano, S.; Colapietro, P.; Cetin, I.; Grati, F.R.; Zanutto, S.; Mandò, C.; Antonazzo, P.; Pileri, P.; Rossella, F.; Larizza, L.; et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth re-striction. Epigenetics 2010, 5, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovina, D.; La Vecchia, M.; Cortesi, A.; Fontana, L.; Pesant, M.; Maitz, S.; Tabano, S.; Bodega, B.; Miozzo, M.; Sirchia, S.M. Profound alterations of the chromatin architecture at chromosome 11p15.5 in cells from Beckwith-Wiedemann and Silver-Russell syndromes patients. Sci. Rep. 2020, 10, 1–19. [Google Scholar] [CrossRef]
- Shu, C.; Zhang, X.; Aouizerat, B.E.; Xu, K. Comparison of methylation capture sequencing and Infinium MethylationEPIC array in peripheral blood mononuclear cells. Epigenetics Chromatin 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Macaulay, E.C.; Weeks, R.J.; Andrews, S.; Morison, I.M. Hypomethylation of functional retrotransposon-derived genes in the human placenta. Mamm. Genome 2011, 22, 722–735. [Google Scholar] [CrossRef]
- Simoni, G.; Sirchia, S.M. Confined placental mosaicism. Prenat. Diagn. 1994, 14, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Updat. 2014, 20, 571–581. [Google Scholar] [CrossRef]
- Decato, B.E.; Lopez-Tello, J.; Sferruzzi-Perri, A.N.; Smith, A.D.; Dean, M.D. DNA Methylation divergence and tissue specialization in the developing mouse placenta. Mol. Biol. Evol. 2017, 34, 1702–1712. [Google Scholar] [CrossRef]
- Paganini, L.; Carlessi, N.; Fontana, L.; Silipigni, R.; Motta, S.; Fiori, S.; Guerneri, S.; Lalatta, F.; Cereda, A.; Sirchia, S.M.; et al. Beckwith–Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics 2015, 10, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Lorincz, M.C.; Schübeler, D. Evidence for converging DNA methylation pathways in placenta and cancer. Dev. Cell 2017, 43, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Calvello, M.; Tabano, S.; Colapietro, P.; Maitz, S.; Pansa, A.; Augello, C.; Lalatta, F.; Gentilin, B.; Spreafico, F.; Calzari, L.; et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beck-with-Wiedemann syndrome. Epigenetics 2013, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight Percentile of Neonates | SGA (n = 33) | AGA (n = 77) | LGA (n = 44) |
|---|---|---|---|
| Maternal age (y) mean ± SD | 35.06 ± 4.53 | 34.79 ± 4.32 | 35.45 ± 3.68 |
| Maternal pregestational weight (kg) mean ± SD | 53.68 ± 6.80 | 59.71 ± 10.37 | 68.32 ± 16.61 |
| Maternal pregestational body mass index (kg/m2) mean ± SD | 20.06 ± 2.21 | 21.85 ± 3.71 | 24.24 ± 5.74 |
| Gestational weight gain (kg) mean ± SD | 9.05 ± 3.25 | 11.17 ± 3.87 | 11.67 ± 4.48 |
| Gestational age at delivery (WOG) mean ± SD | 38 ± 2 | 39 ± 2 | 39 ± 2 |
| Mode of delivery (Vaginal Birth/ Cesarean Section) count; percentage | VB (n = 13; 8.44%) CS (n = 20; 12.99%) | VB (n = 24; 15.60%) CS (n = 53; 34.42%) | VB (n = 18; 11.69%) CS (n = 26; 16.88%) |
| Birthweight (g) mean ± SD | 2486.67 ± 335.61 | 3360.91 ± 434.23 | 4109.77 ± 258.19 |
| Placental weight (g) mean ± SD | 475.19 ± 98.97 | 587.95 ± 97.12 | 741.47 ± 145.38 |
| A | |
|---|---|
| Diseases or Functions Annotation | p-Value |
| Leukocyte migration | 1.52 × 10−16 |
| Cell movement of lymphocytes | 5.05 × 10−14 |
| Cell movement of mononuclear leukocytes | 6.30 × 10−14 |
| Quantity of leukocytes | 7.50 × 10−14 |
| Lymphocyte migration | 9.59 × 10−14 |
| Quantity of lymphatic system cells | 1.97 × 10−13 |
| Quantity of lymphocytes | 5.29 × 10−13 |
| Cell movement of leukocytes | 6.80 × 10−13 |
| Proliferation of blood cells | 1.21 × 10−12 |
| Proliferation of immune cells | 1.62 × 10−12 |
| B | |
| Diseases or Functions Annotation | p-Value |
| Cutaneous melanoma | 1.52 × 10−12 |
| Skin tumor | 5.94 × 10−12 |
| Melanoma | 1.01 × 10−11 |
| Skin cancer | 1.20 × 10−11 |
| Malignant neuroendocrine neoplasm | 2.45 × 10−7 |
| Small cell lung carcinoma | 3.07 × 10−7 |
| Olfactory response | 3.73 × 10−7 |
| Neuroendocrine tumor | 4.48 × 10−7 |
| Extrapancreatic neuroendocrine tumor | 5.84 × 10−7 |
| Blue round small cell tumor | 7.16 × 10−6 |
| A | ||
|---|---|---|
| Diseases or Functions Annotation | p-Value (Microarray) | p-Value (Methyl-Seq) |
| Leukocyte migration | 2.93 × 10−26 | 4.53 × 10−7 |
| Quantity of lymphatic system cells | 3.30 × 10−25 | 4.34 × 10−6 |
| Quantity of leukocytes | 1.51 × 10−25 | 3.42 × 10−6 |
| Quantity of lymphocytes | 1.53 × 10−24 | 4.75 × 10−6 |
| Cell movement of leukocytes | 5.69 × 10−23 | 1.38 × 10−10 |
| Activation of cells | 2.86 × 10−22 | 5.05 × 10−14 |
| Migration of cells | 1.08 × 10−21 | 7.35 × 10−7 |
| Proliferation of lymphatic system cells | 1.68 × 10−21 | 1.62 × 10−6 |
| Proliferation of lymphocytes | 5.96 × 10−21 | 2.09 × 10−6 |
| Proliferation of immune cells | 3.46 × 10−20 | 1.58 × 10−6 |
| B | ||
| Diseases or Functions Annotation | p-Value (Microarray) | p-Value (Methyl-Seq) |
| Cross-linkage of protein | 1.60 × 10−4 | 8.28 × 10−4 |
| Skin carcinoma | 7.37 × 10−4 | 3.95 × 10−3 |
| Skin squamous cell carcinoma | 9.61 × 10−4 | 2.69 × 10−4 |
| Relaxation of heart ventricle | 1.03 × 10−3 | 5.24 × 10−3 |
| Surface area of ventricular myocytes | 1.22 × 10−3 | 6.15 × 10−3 |
| Gastro-esophageal carcinoma | 3.77 × 10−3 | 2.78 × 10−3 |
| Chemosensitivity of glioblastoma cells | 4.03 × 10−3 | 9.20 × 10−3 |
| Pancreatic lesion | 5.88 × 10−3 | 6.34 × 10−3 |
| Formation of solid tumor | 5.90 × 10−3 | 1.74 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rondinone, O.; Murgia, A.; Costanza, J.; Tabano, S.; Camanni, M.; Corsaro, L.; Fontana, L.; Colapietro, P.; Calzari, L.; Motta, S.; et al. Extensive Placental Methylation Profiling in Normal Pregnancies. Int. J. Mol. Sci. 2021, 22, 2136. https://doi.org/10.3390/ijms22042136
Rondinone O, Murgia A, Costanza J, Tabano S, Camanni M, Corsaro L, Fontana L, Colapietro P, Calzari L, Motta S, et al. Extensive Placental Methylation Profiling in Normal Pregnancies. International Journal of Molecular Sciences. 2021; 22(4):2136. https://doi.org/10.3390/ijms22042136
Chicago/Turabian StyleRondinone, Ornella, Alessio Murgia, Jole Costanza, Silvia Tabano, Margherita Camanni, Luigi Corsaro, Laura Fontana, Patrizia Colapietro, Luciano Calzari, Silvia Motta, and et al. 2021. "Extensive Placental Methylation Profiling in Normal Pregnancies" International Journal of Molecular Sciences 22, no. 4: 2136. https://doi.org/10.3390/ijms22042136
APA StyleRondinone, O., Murgia, A., Costanza, J., Tabano, S., Camanni, M., Corsaro, L., Fontana, L., Colapietro, P., Calzari, L., Motta, S., Santaniello, C., Radaelli, T., Ferrazzi, E., Bosari, S., Gentilini, D., Sirchia, S. M., & Miozzo, M. (2021). Extensive Placental Methylation Profiling in Normal Pregnancies. International Journal of Molecular Sciences, 22(4), 2136. https://doi.org/10.3390/ijms22042136