
Time-Resolved scRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment

 , ,
, , 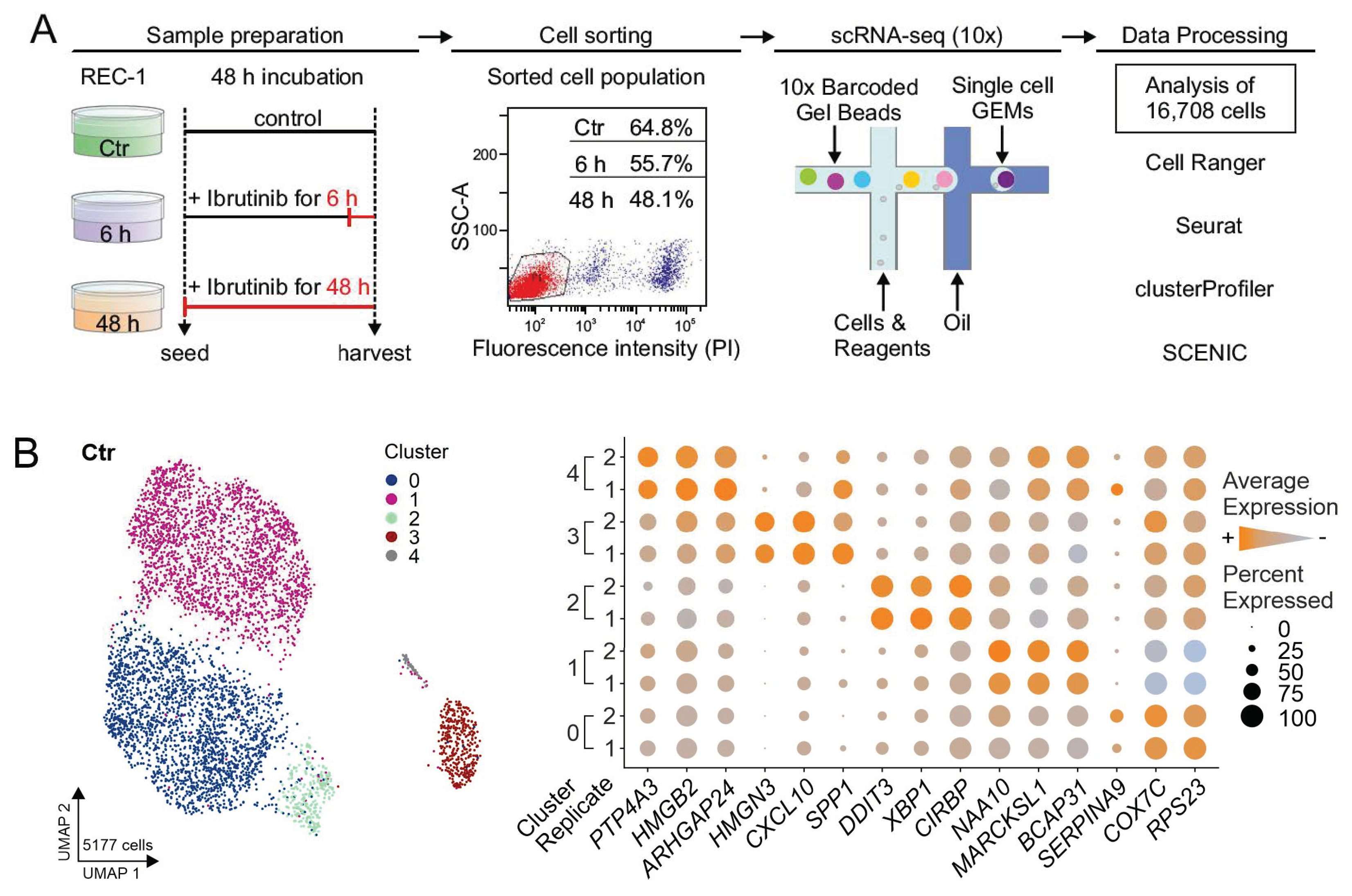{kind=link}
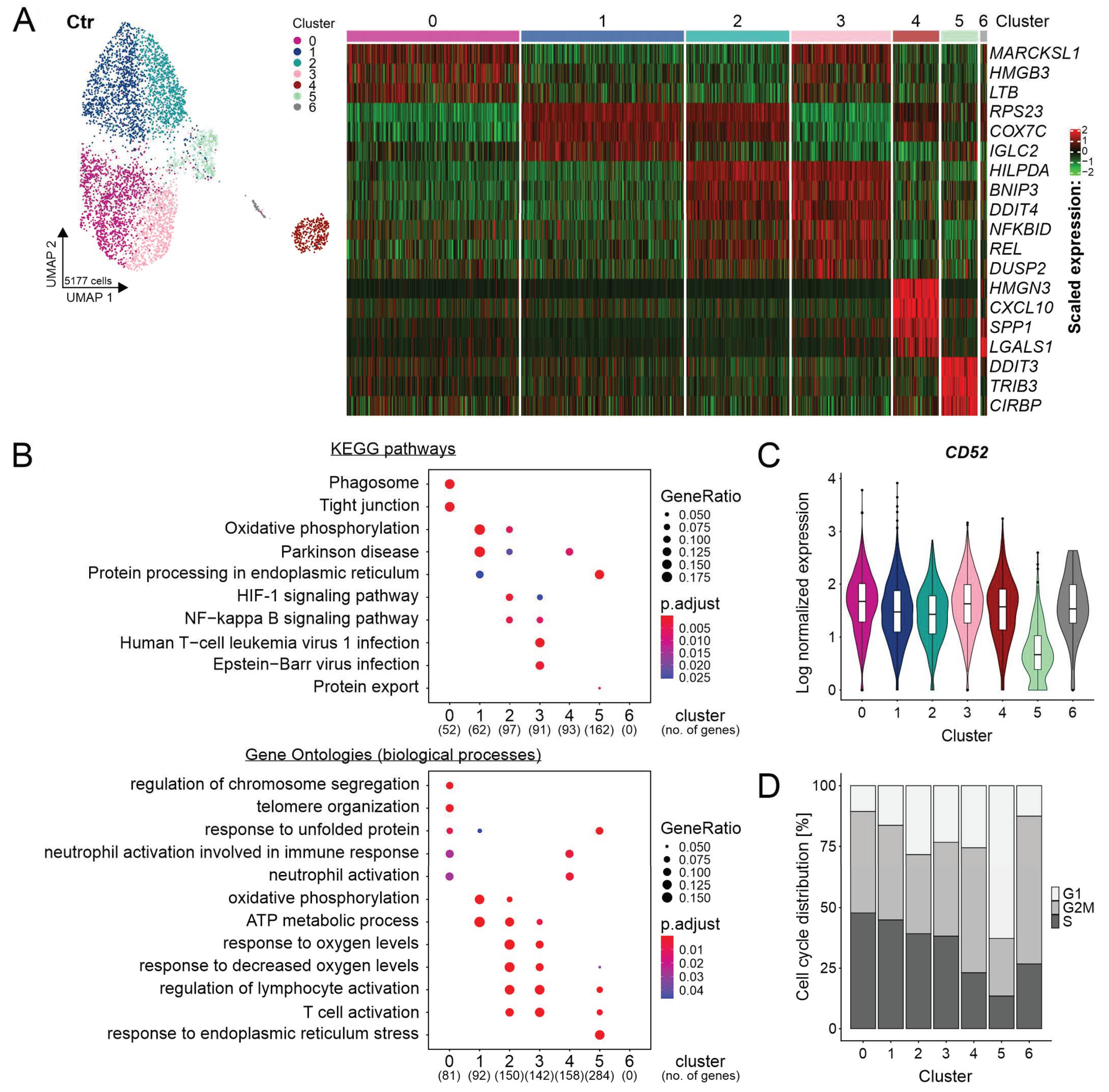{kind=link}
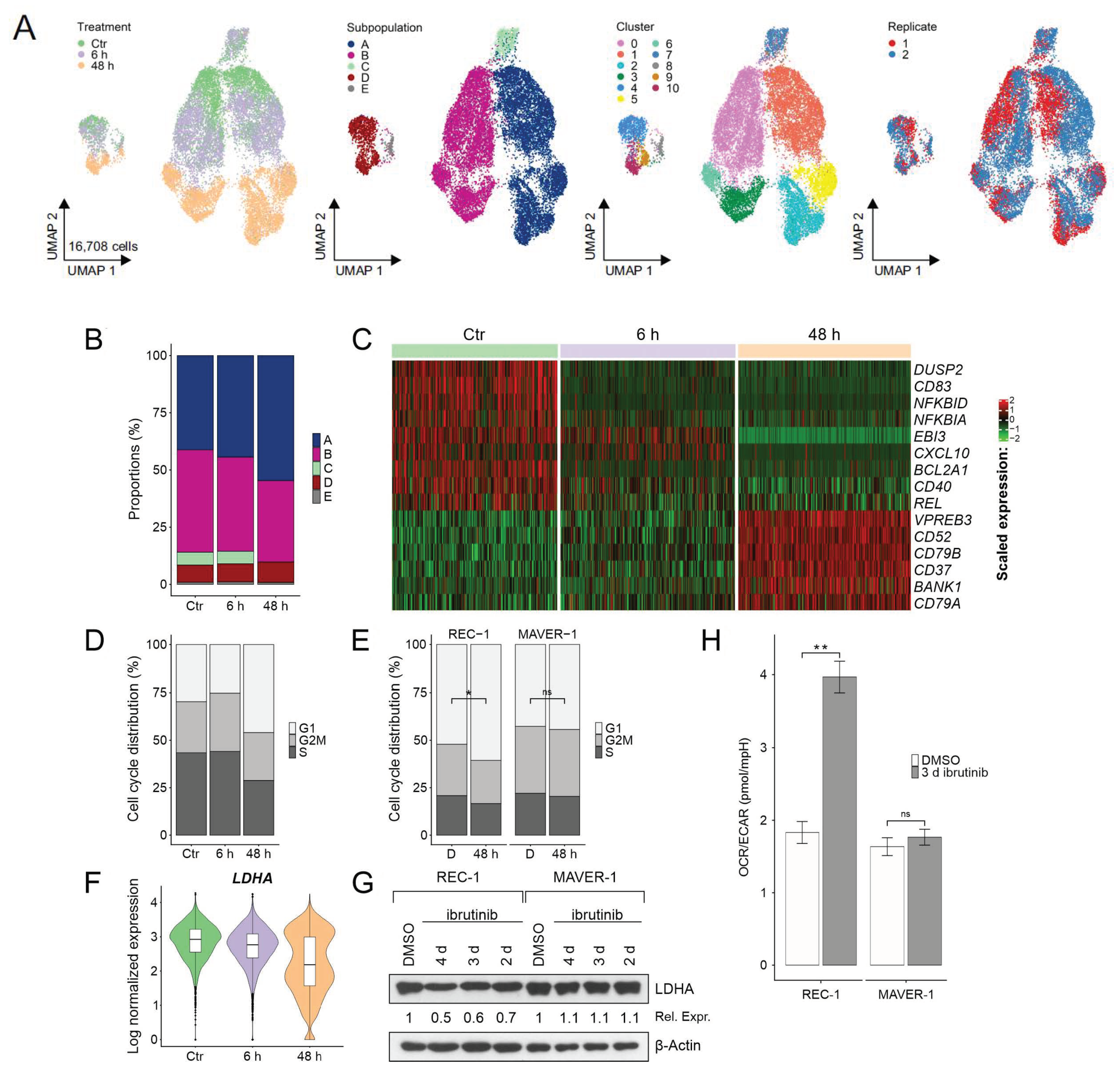{kind=link}
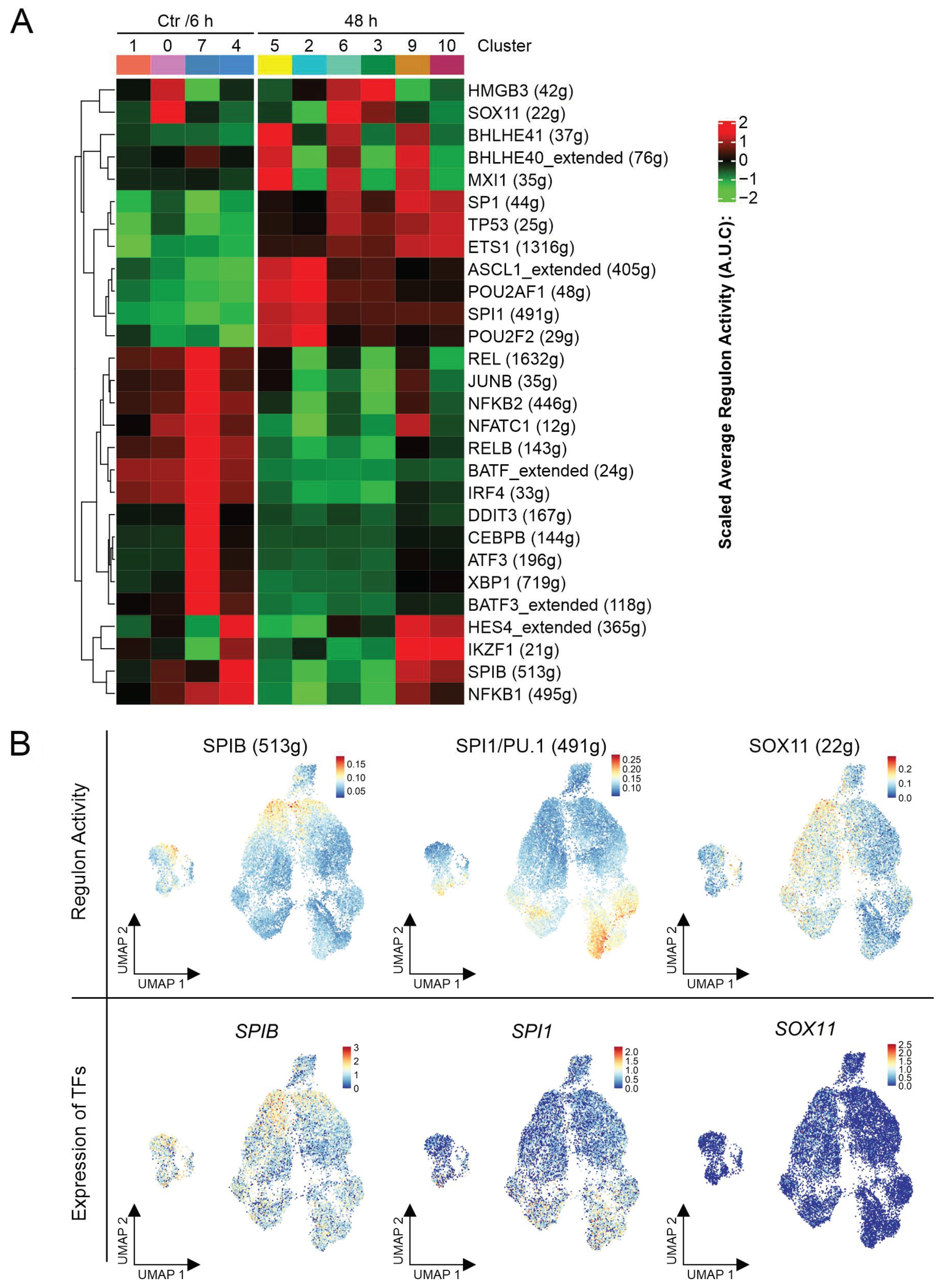{kind=link}
Abstract
:1. Introduction
2. Results
2.1. High Reproducibility of Replicates
2.2. Heterogeneity of REC-1
2.3. Evolution and Common Response of Subpopulations
2.4. Gene Regulatory Networks during Ibrutinib Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Single-Cell RNA Sequencing
4.2.1. Experiment Set-Up
4.2.2. Data Analysis
4.2.3. Batch Correction and Cell Cycle Regression
4.2.4. ClusterProfiler
4.2.5. SCENIC Analysis
4.3. Cell Cycle Analysis
4.4. Western Blot
4.5. Extracellular Flux Analysis
4.6. Flow Cytometric Analysis of Cell-Surface Antigens
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, P.; Wang, M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am. J. Hematol. 2019, 94, 710–725. [Google Scholar] [CrossRef] [Green Version]
- Mozos, A.; Royo, C.; Hartmann, E.; De Jong, D.; Baro, C.; Valera, A.; Fu, K.; Weisenburger, D.D.; Delabie, J.; Chuang, S.S.; et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009, 94, 1555–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.E.; Swerdlow, S.H. Cyclin D1 overexpression in non-Hodgkin’s lymphoma with chromosome 11 bcl-1 rearrangement. Ann. Oncol. 1994, 5 (Suppl. 1), 71–73. [Google Scholar] [CrossRef]
- Fernandez, V.; Salamero, O.; Espinet, B.; Sole, F.; Royo, C.; Navarro, A.; Camacho, F.; Bea, S.; Hartmann, E.; Amador, V.; et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010, 70, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rule, S. The modern approach to mantle cell lymphoma. Hematol. Oncol. 2019, 37 (Suppl. 1), 66–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, L.; Gafter-Gvili, A.; Dreyling, M.; Ghielmini, M.; Witzens-Harig, M.; Shpilberg, O.; Unterhalt, M.; Rummel, M.; Gurion, R. Maintenance Treatment for Patients With Mantle Cell Lymphoma: A Systematic Review and Meta-analysis of Randomized Trials. Hemasphere 2018, 2, e136. [Google Scholar] [CrossRef]
- Maddocks, K. Update on mantle cell lymphoma. Blood 2018, 132, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Ribrag, V. Novel agents for mantle cell lymphoma: Molecular rational and clinical data. Expert Opin. Investig. Drugs 2020, 29, 555–566. [Google Scholar] [CrossRef]
- Fichtner, M.; Dreyling, M.; Binder, M.; Trepel, M. The role of B cell antigen receptors in mantle cell lymphoma. J. Hematol. Oncol. 2017, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chan, F.C.; Meissner, B.; Bhang, H.E.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 2014, 20, 87–92. [Google Scholar] [CrossRef]
- Pham, L.V.; Tamayo, A.T.; Yoshimura, L.C.; Lo, P.; Ford, R.J. Inhibition of Constitutive NF-κB Activation in Mantle Cell Lymphoma B Cells Leads to Induction of Cell Cycle Arrest and Apoptosis. J. Immunol. 2003, 171, 88–95. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Hershkovitz-Rokah, O.; Pulver, D.; Lenz, G.; Shpilberg, O. Ibrutinib resistance in mantle cell lymphoma: Clinical, molecular and treatment aspects. Br. J. Haematol. 2018, 181, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mato Prado, M.; Frampton, A.E.; Stebbing, J.; Krell, J. Single-cell sequencing in cancer research. Expert Rev. Mol. Diagn. 2015, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Roider, T.; Seufert, J.; Uvarovskii, A.; Frauhammer, F.; Bordas, M.; Abedpour, N.; Stolarczyk, M.; Mallm, J.-P.; Herbst, S.A.; Bruch, P.-M.; et al. Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels. Nat. Cell Biol. 2020, 22, 896–906. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Mo, S.; Li, X.; He, Y.; Yang, J. Single-cell RNA-seq reveals the immune escape and drug resistance mechanisms of mantle cell lymphoma. Cancer Biol. Med. 2020, 17, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Rauert-Wunderlich, H.; Rudelius, M.; Ott, G.; Rosenwald, A. Targeting protein kinase C in mantle cell lymphoma. Br. J. Haematol. 2016, 173, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Has Significant Activity in Patients with Relapsed/Refractory B-Cell Malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Ma, J.; Lu, P.; Guo, A.; Cheng, S.; Zong, H.; Martin, P.; Coleman, M.; Wang, Y.L. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br. J. Haematol. 2014, 166, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Abramson, J.S.; Pinkus, G.S.; Treon, S.P.; Dorfman, D.M.; Dong, H.Y.; Shipp, M.A.; Kutok, J.L. Heterogeneous CD52 expression among hematologic neoplasms: Implications for the use of alemtuzumab (CAMPATH-1H). Clin. Cancer Res. 2006, 12, 7174–7179. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Asplund, A.C.; Porwit, A.; Flygare, J.; Smith, C.I.; Christensson, B.; Sander, B. The subcellular Sox11 distribution pattern identifies subsets of mantle cell lymphoma: Correlation to overall survival. Br. J. Haematol. 2008, 143, 248–252. [Google Scholar] [CrossRef]
- Chiron, D.; Di Liberto, M.; Martin, P.; Huang, X.; Sharman, J.; Blecua, P.; Mathew, S.; Vijay, P.; Eng, K.; Ali, S.; et al. Cell-Cycle Reprogramming for PI3K Inhibition Overrides a Relapse-Specific C481S BTK Mutation Revealed by Longitudinal Functional Genomics in Mantle Cell Lymphoma. Cancer Discov. 2014, 4, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shaffer, A.L., III; Emre, N.C.T.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.N.; Tellier, J.; Liao, Y.; Trezise, S.; Light, A.; O’Donnell, K.; Garrett-Sinha, L.A.; Shi, W.; Tarlinton, D.M.; Nutt, S.L. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat. Commun. 2017, 8, 1426. [Google Scholar] [CrossRef]
- Solomon, L.A.; Li, S.K.; Piskorz, J.; Xu, L.S.; DeKoter, R.P. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genom. 2015, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Pagel, J.M.; Spurgeon, S.E.; Byrd, J.C.; Awan, F.T.; Flinn, I.W.; Lanasa, M.C.; Eisenfeld, A.J.; Stromatt, S.C.; Gopal, A.K. Otlertuzumab (TRU-016), an anti-CD37 monospecific ADAPTIR™therapeutic protein, for relapsed or refractory NHL patients. Br. J. Haematol. 2015, 168, 38–45. [Google Scholar] [CrossRef]
- Hinz, M.; Löser, P.; Mathas, S.; Krappmann, D.; Dörken, B.; Scheidereit, C. Constitutive NF-κB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood 2001, 97, 2798–2807. [Google Scholar] [CrossRef]
- Rendeiro, A.F.; Krausgruber, T.; Fortelny, N.; Zhao, F.; Penz, T.; Farlik, M.; Schuster, L.C.; Nemc, A.; Tasnady, S.; Reti, M.; et al. Chromatin mapping and single-cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nat. Commun. 2020, 11, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinker, G.S.; Greenwald, A.C.; Tal, R.; Orlova, Z.; Cuoco, M.S.; McFarland, J.M.; Warren, A.; Rodman, C.; Roth, J.A.; Bender, S.A.; et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020, 52, 1208–1218. [Google Scholar] [CrossRef]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of Biomarkers Predictive of GSI Response in Triple-Negative Breast Cancer and Adenoid Cystic Carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Silkenstedt, E.; Arenas, F.; Colom-Sanmarti, B.; Xargay-Torrent, S.; Higashi, M.; Giro, A.; Rodriguez, V.; Fuentes, P.; Aulitzky, W.E.; van der Kuip, H.; et al. Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy. J. Exp. Clin. Cancer Res. 2019, 38, 446. [Google Scholar] [CrossRef] [Green Version]
- Wightman, S.C.; Uppal, A.; Pitroda, S.P.; Ganai, S.; Burnette, B.; Stack, M.; Oshima, G.; Khan, S.; Huang, X.; Posner, M.C.; et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br. J. Cancer 2015, 113, 327–335. [Google Scholar] [CrossRef]
- Yushi, Q.; Li, Z.; Von Roemeling, C.A.; Doeppler, H.; Marlow, L.A.; Kim, B.Y.; Radisky, D.C.; Storz, P.; Copland, J.A.; Tun, H.W. Osteopontin is a multi-faceted pro-tumorigenic driver for central nervous system lymphoma. Oncotarget 2016, 7, 32156–32171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevde, L.A.; Samant, R.S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 2014, 37, 131–141. [Google Scholar] [CrossRef]
- Anborgh, P.H.; Mutrie, J.C.; Tuck, A.B.; Chambers, A.F. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J. Cell Mol. Med. 2010, 14, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Storti, P.; Marchica, V.; Giuliani, N. Role of Galectins in Multiple Myeloma. Int. J. Mol. Sci. 2017, 18, 2740. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Ford, R.J. The microenvironment in mantle cell lymphoma: Cellular and molecular pathways and emerging targeted therapies. Semin. Cancer Biol. 2011, 21, 308–312. [Google Scholar] [CrossRef]
- Faderl, S.; Thomas, D.A.; O’Brien, S.; Garcia-Manero, G.; Kantarjian, H.M.; Giles, F.J.; Koller, C.; Ferrajoli, A.; Verstovsek, S.; Pro, B.; et al. Experience with alemtuzumab plus rituximab in patients with relapsed and refractory lymphoid malignancies. Blood 2003, 101, 3413–3415. [Google Scholar] [CrossRef]
- Keating, M.J.; Flinn, I.; Jain, V.; Binet, J.-L.; Hillmen, P.; Byrd, J.; Albitar, M.; Brettman, L.; Santabarbara, P.; Wacker, B.; et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: Results of a large international study. Blood 2002, 99, 3554–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winqvist, M.; Palma, M.; Heimersson, K.; Mellstedt, H.; Österborg, A.; Lundin, J. Dual targeting of Bruton tyrosine kinase and CD52 induces minimal residual disease-Negativity in the bone marrow of poor-prognosis chronic lymphocytic leukaemia patients but is associated with opportunistic infections-Results from a phase I study. Br. J. Haematol. 2018, 182, 590–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11, eaau1167. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.H.; Pervolarakis, N.; Nee, K.; Kessenbrock, K. Experimental Considerations for Single-Cell RNA Sequencing Approaches. Front. Cell Dev. Biol. 2018, 6, 108. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer, D.; Campo, E. Unlocking new therapeutic targets and resistance mechanisms in mantle cell lymphoma. Cancer Cell 2014, 25, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Rauert-Wunderlich, H.; Rudelius, M.; Berberich, I.; Rosenwald, A. CD40L mediated alternative NFkappaB-signaling induces resistance to BCR-inhibitors in patients with mantle cell lymphoma. Cell Death Dis. 2018, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Göttgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuhr, V.; Vafadarnejad, E.; Dietrich, O.; Arampatzi, P.; Riedel, A.; Saliba, A.-E.; Rosenwald, A.; Rauert-Wunderlich, H. Time-Resolved scRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment. Int. J. Mol. Sci. 2021, 22, 2276. https://doi.org/10.3390/ijms22052276
Fuhr V, Vafadarnejad E, Dietrich O, Arampatzi P, Riedel A, Saliba A-E, Rosenwald A, Rauert-Wunderlich H. Time-Resolved scRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment. International Journal of Molecular Sciences. 2021; 22(5):2276. https://doi.org/10.3390/ijms22052276
Chicago/Turabian StyleFuhr, Viktoria, Ehsan Vafadarnejad, Oliver Dietrich, Panagiota Arampatzi, Angela Riedel, Antoine-Emmanuel Saliba, Andreas Rosenwald, and Hilka Rauert-Wunderlich. 2021. "Time-Resolved scRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment" International Journal of Molecular Sciences 22, no. 5: 2276. https://doi.org/10.3390/ijms22052276
APA StyleFuhr, V., Vafadarnejad, E., Dietrich, O., Arampatzi, P., Riedel, A., Saliba, A. -E., Rosenwald, A., & Rauert-Wunderlich, H. (2021). Time-Resolved scRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment. International Journal of Molecular Sciences, 22(5), 2276. https://doi.org/10.3390/ijms22052276