
Comparing Ligninolytic Capabilities of Bacterial and Fungal Dye-Decolorizing Peroxidases and Class-II Peroxidase-Catalases

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Peroxidase Production
2.2. Kinetics for Oxidation of Simple Peroxidase Substrates
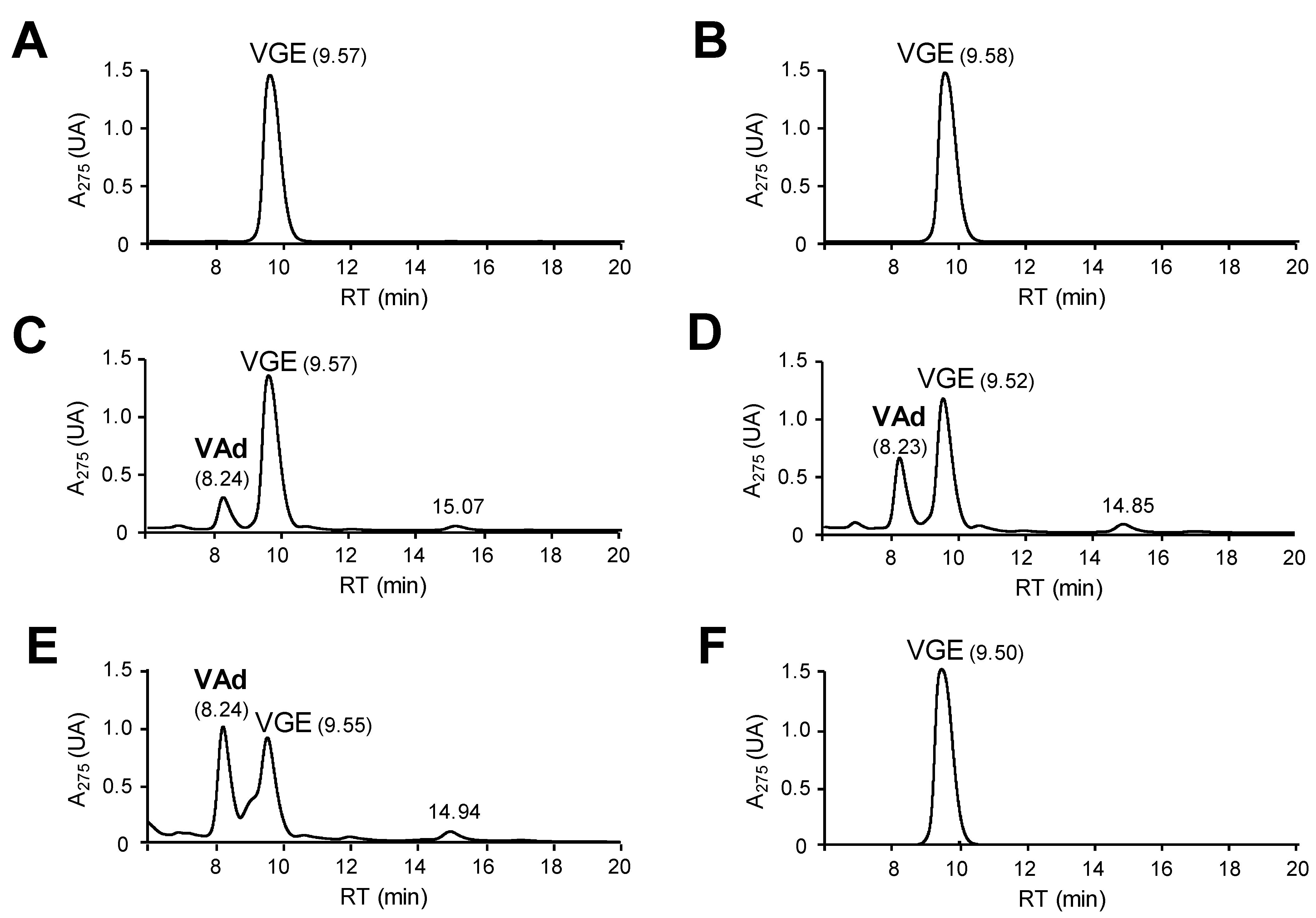
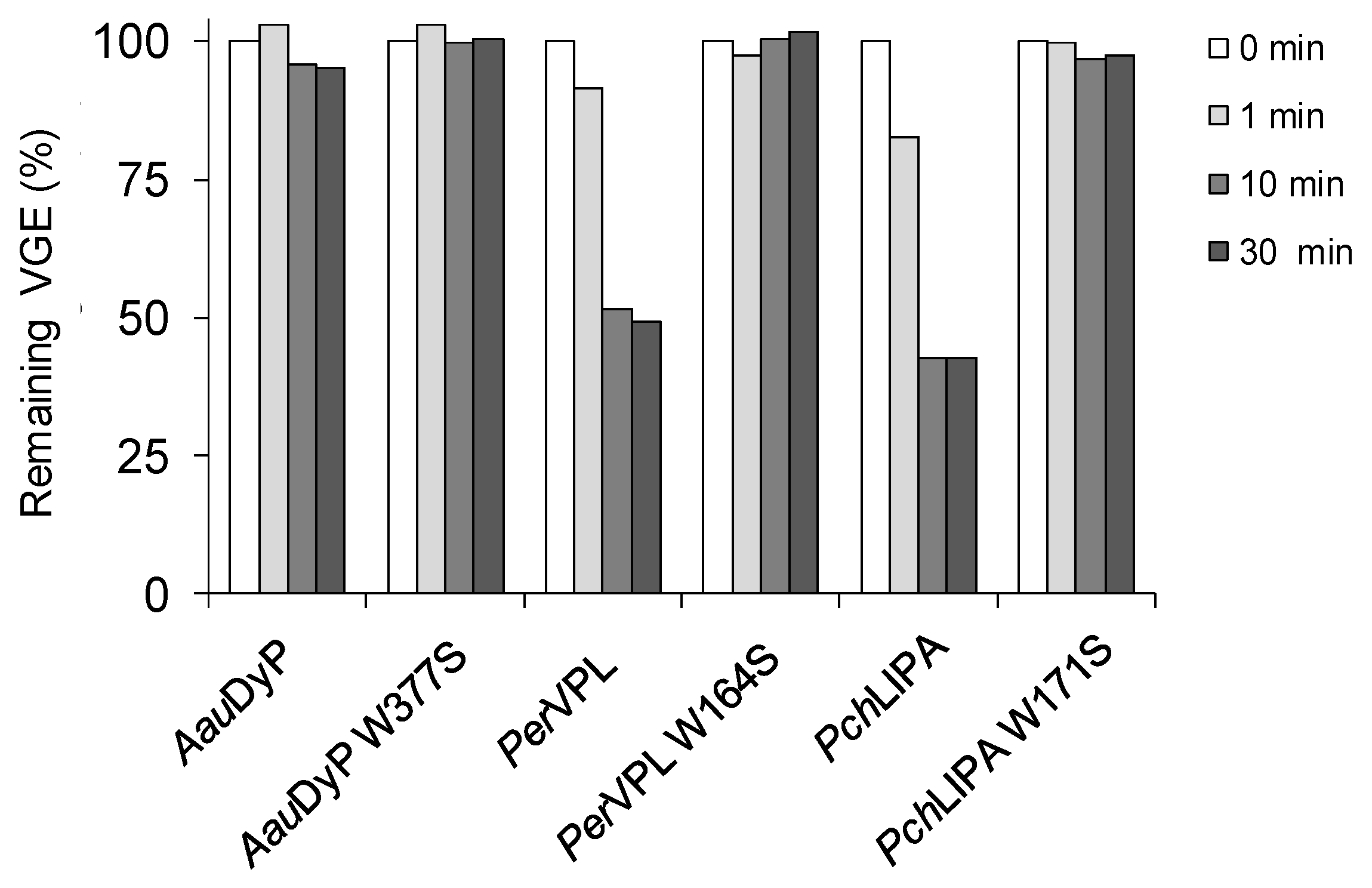
2.3. Oxidation of Lignin Model Dimers
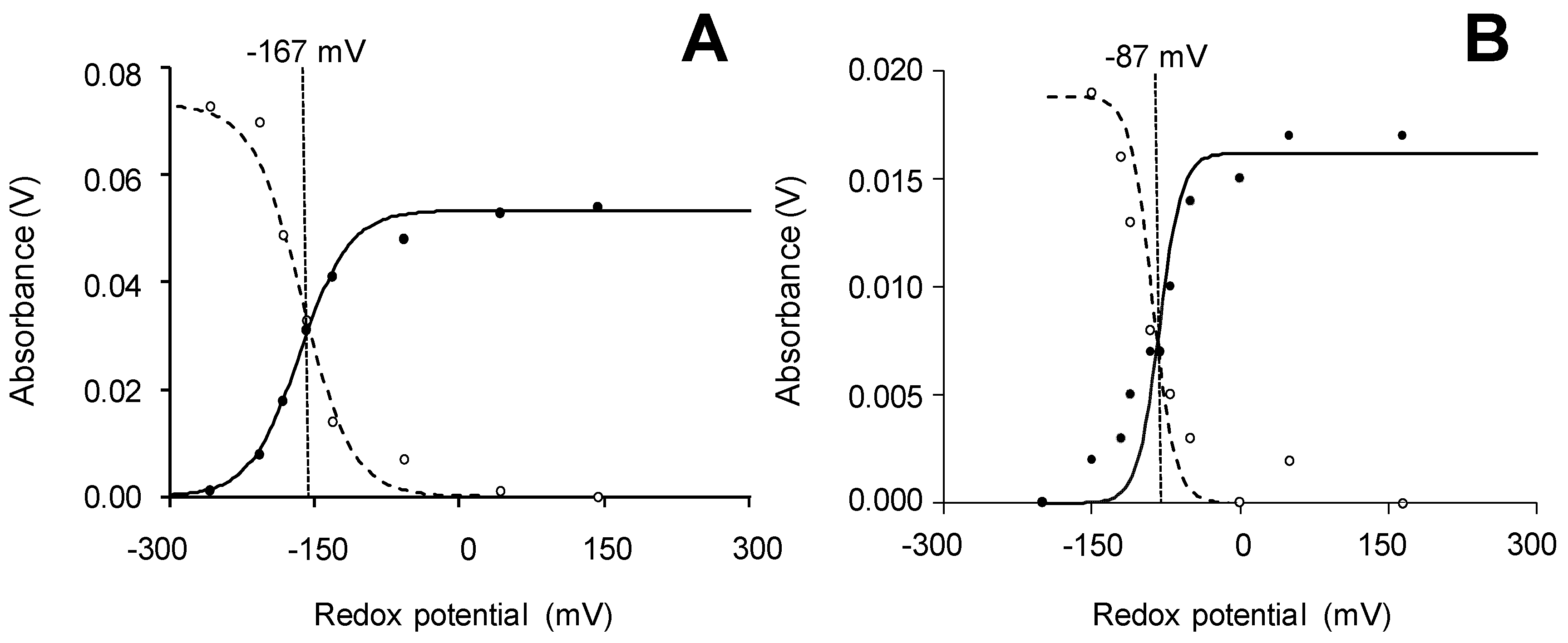
2.4. Reduction-Potential (Eo′) Measurements
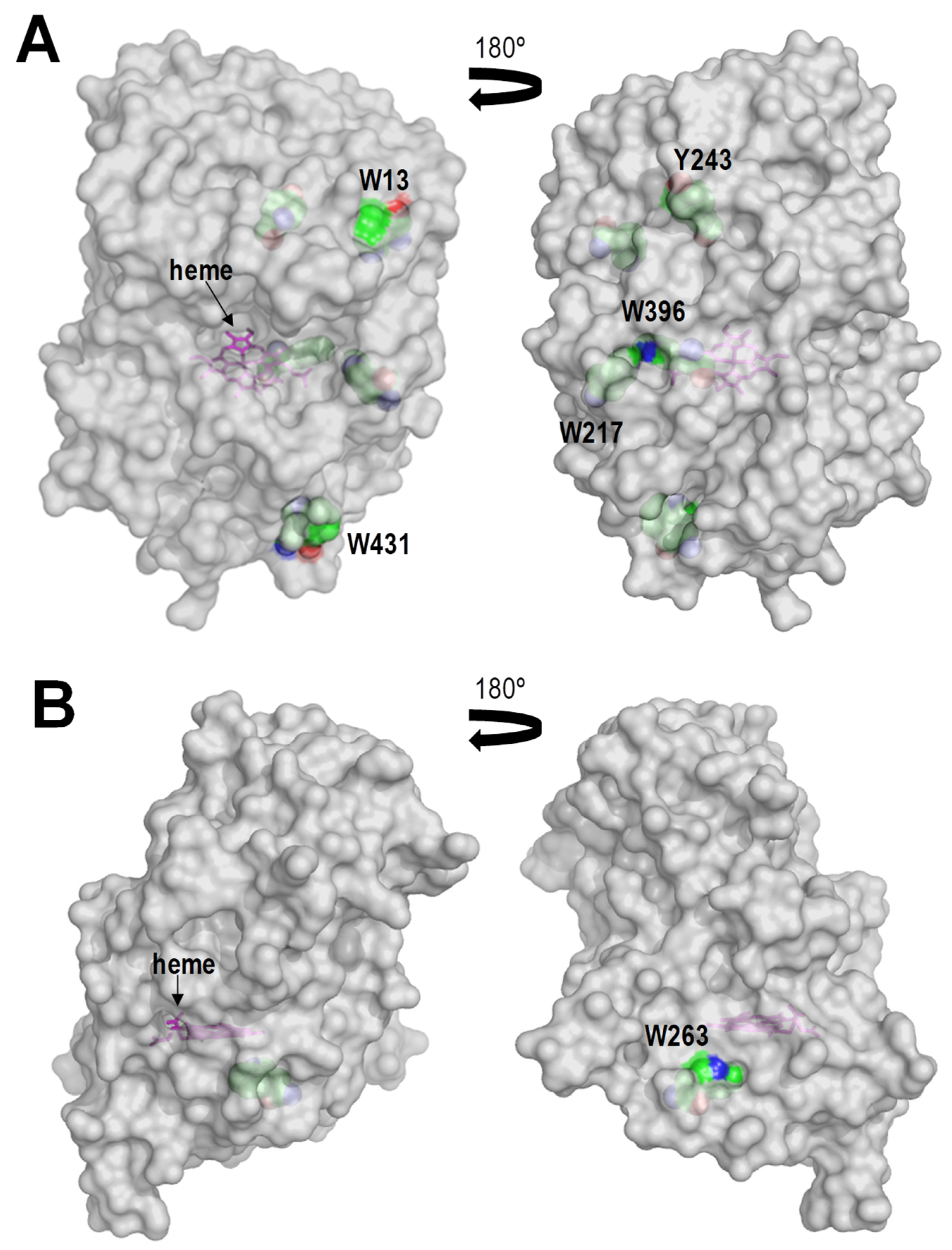
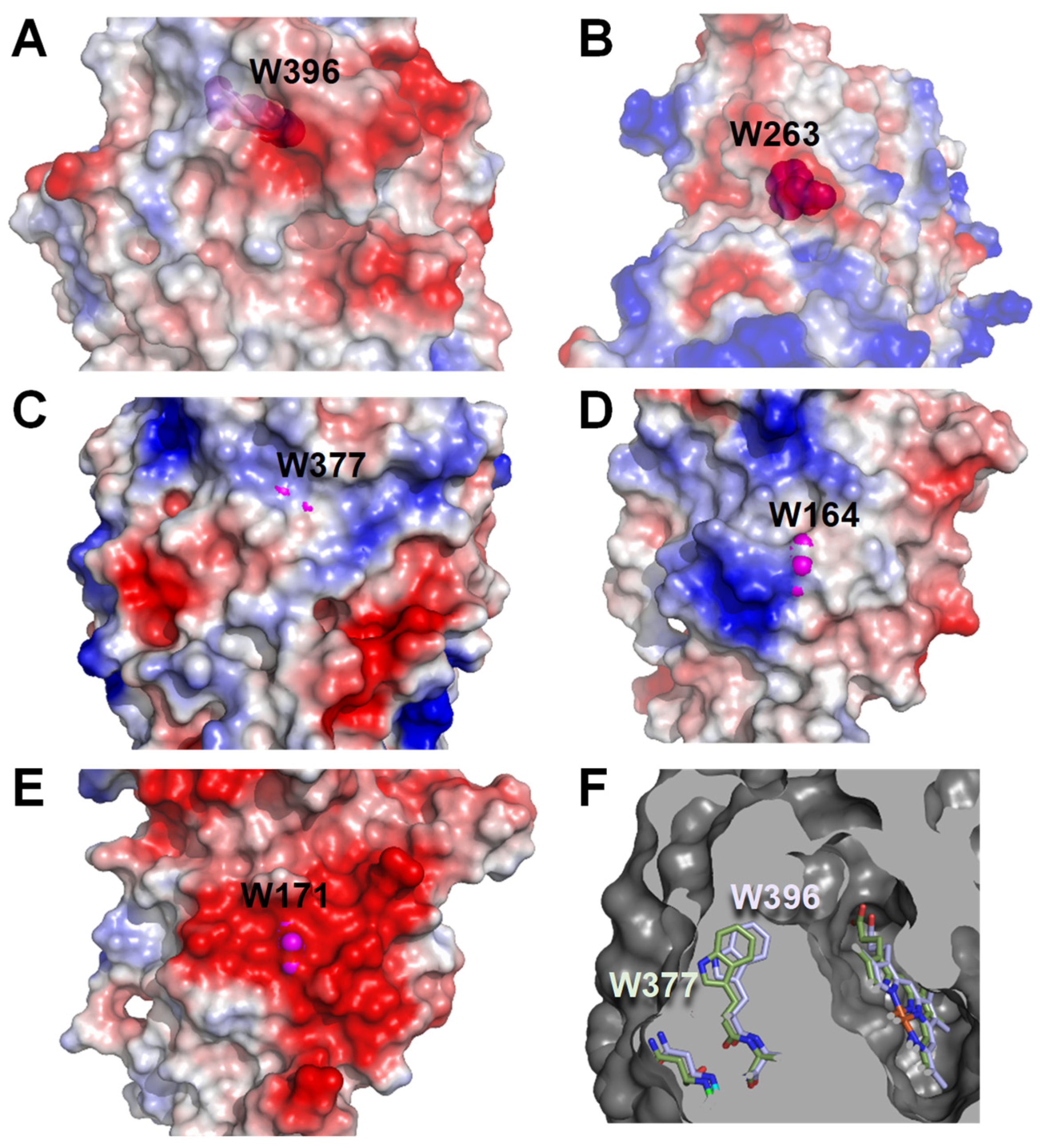
2.5. Surface Features of the Bacterial and Fungal Peroxidases Analyzed
2.6. Overview on Relevant Characteristics of Bacterial and Fungal DyPs
3. Materials and Methods
3.1. Chemicals
3.2. Enzymes Expression, Purification and nLC-MS/MS Analysis
3.3. Enzyme Kinetics
3.4. Chromatographic Analysis of Model Dimer Oxidation
3.5. Spectro-Electrochemical Measurement of Fe3+/Fe2+ Redox Potentials
3.6. Stopped-Flow Estimation of Eo′ Values of Catalytic-Cycle Intermediates
3.7. Structural Analysis of the Different Peroxidases
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanholme, R.; De Meester, B.; Ralph, J.; Boerjan, W. Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 2019, 56, 230–239. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Martínez, M.J.; del Río, J.C.; Gutiérrez, A. Enzymatic delignification of plant cell wall: From nature to mill. Curr. Opin. Biotechnol. 2009, 20, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Bajwa, D.S.; Pourhashem, G.; Ullah, A.H.; Bajwa, S.G. A concise review of current lignin production, applications, products and their environmental impact. Ind. Crops Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Beckham, G.T. Lignin Valorization: Emerging Approaches; RSC: Cambridge, UK, 2018. [Google Scholar]
- Zhang, Y.H.P. Reviving the carbohydrate economy via multi-product lignocellulose biorefineries. J. Ind. Microbiol. Biotechnol. 2008, 35, 367–375. [Google Scholar] [CrossRef]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef] [Green Version]
- Martínez, A.T.; Camarero, S.; Ruiz-Dueñas, F.J.; Martínez, M.J. Biological lignin degradation. In Lignin Valorization: Emerging Approaches; Beckham, G.T., Ed.; RSC: Cambridge, UK, 2018; pp. 199–225. [Google Scholar]
- Zámocký, M.; Hofbauer, S.; Schaffner, I.; Gasselhuber, B.; Nicolussi, A.; Soudi, M.; Pirker, K.F.; Furtmüller, P.G.; Obinger, C. Independent evolution of four heme peroxidase superfamilies. Arch. Biochem. Biophys. 2015, 574, 108–119. [Google Scholar] [CrossRef]
- Cavener, D.R. GMC oxidoreductases. A newly defined family of homologous proteins with diverse catalytic activities. J. Mol. Biol. 1992, 223, 811–814. [Google Scholar] [CrossRef]
- Vanden Wymelenberg, A.; Gaskell, J.; Mozuch, M.; Sabat, G.; Ralph, J.; Skyba, O.; Mansfield, S.D.; Blanchette, R.A.; Martinez, D.; Grigoriev, I.; et al. Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium. Appl. Environ. Microbiol. 2010, 76, 3599–3610. [Google Scholar] [CrossRef] [Green Version]
- Vanden Wymelenberg, A.; Sabat, G.; Mozuch, M.; Kersten, P.J.; Cullen, D.; Blanchette, R.A. Structure, organization, and transcriptional regulation of a family of copper radical oxidase genes in the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl. Environ. Microbiol. 2006, 72, 4871–4877. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Go, N. Function and molecular evolution of multicopper blue proteins. Cell. Mol. Life Sci. 2005, 62, 2050–2066. [Google Scholar] [CrossRef]
- Crawford, D.L.; Pometto, A.L., III; Crawford, R.L. Lignin degradation by Streptomyces viridosporus: Isolation and characterization of a new polymeric lignin degradation intermediate. Appl. Environ. Microbiol. 1983, 45, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granja-Travez, R.S.; Persinoti, G.F.; Squina, F.M.; Bugg, T.D.H. Functional genomic analysis of bacterial lignin degraders: Diversity in mechanisms of lignin oxidation and metabolism. Appl. Microbiol. Biotechnol. 2020, 104, 3305–3320. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Singh, R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 2011, 22, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.X.; Lei, P.; Zhai, R.; Wen, Z.Q.; Jin, M.J. Recent advances in lignin valorization with bacterial cultures: Microorganisms, metabolic pathways, and bio-products. Biotechnol. Biofuels 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Chang, M.C.Y. Exploring bacterial lignin degradation. Curr. Opin. Chem. Biol. 2014, 19, 1–7. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Colpa, D.I.; Habib, M.H.M.; Fraaije, M.W. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.X.; Yue, F.F.; Cui, Y.L.; Xu, Y.M.; Shan, Y.Y.; Liu, B.F.; Zhou, Y.; Lu, X. Biodegradation of lignin by Pseudomonas sp. Q18 and the characterization of a novel bacterial DyP-type peroxidase. J. Ind. Microbiol. Biotechnol. 2018, 45, 913–927. [Google Scholar] [CrossRef]
- Rashid, G.M.M.; Zhang, X.Y.; Wilkinson, R.C.; Fulop, V.; Cottyn, B.; Baumberger, S.; Bugg, T.D.H. Sphingobacterium sp. T2 manganese superoxide dismutase catalyzes the oxidative demethylation of polymeric lignin via generation of hydroxyl radical. ACS Chem. Biol. 2018, 13, 2920–2929. [Google Scholar] [CrossRef]
- Chandra, R.; Kumar, V.; Yadav, S. Extremophilic ligninolytic enzymes. In Extremophilic Enzymatic Processing of Lignocellulosic Feedstocks to Bioenergy; Sani, R.K., Krishnaraj, R.N., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 115–154. [Google Scholar]
- Rai, R.; Bibra, M.; Chadha, B.S.; Sani, R.K. Enhanced hydrolysis of lignocellulosic biomass with doping of a highly thermostable recombinant laccase. Int. J. Biol. Macromol. 2019, 137, 232–237. [Google Scholar] [CrossRef]
- Liers, C.; Pecyna, M.J.; Kellner, H.; Worrich, A.; Zorn, H.; Steffen, K.T.; Hofrichter, M.; Ullrich, R. Substrate oxidation by dye-decolorizing peroxidases (DyPs) from wood- and litter-degrading agaricomycetes compared to other fungal and plant heme-peroxidases. Appl. Microbiol. Biotechnol. 2013, 87, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Linde, D.; Coscolín, C.; Liers, C.; Hofrichter, M.; Martínez, A.T.; Ruiz-Dueñas, F.J. Heterologous expression and physicochemical characterization of a fungal dye-decolorizing peroxidase from Auricularia auricula-judae. Protein Express. Purif. 2014, 103, 28–37. [Google Scholar] [CrossRef]
- Linde, D.; Ruiz-Dueñas, F.J.; Fernández-Fueyo, E.; Guallar, V.; Hammel, K.E.; Pogni, R.; Martínez, A.T. Basidiomycete DyPs: Genomic diversity, structural-functional aspects, reaction mechanism and environmental significance. Arch. Biochem. Biophys. 2015, 574, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Gong, G.; Woo, H.M.; Kim, Y.; Um, Y. A dye-decolorizing peroxidase from Bacillus subtilis exhibiting substrate-dependent optimum temperature for dyes and β-ether lignin dimer. Sci. Rep. 2015, 5, 8245. [Google Scholar] [CrossRef]
- Ogola, H.J.O.; Kamiike, T.; Hashimoto, N.; Ashida, H.; Ishikawa, T.; Shibata, H.; Sawa, Y. Molecular characterization of a novel peroxidase from the cyanobacterium Anabaena sp strain PCC 7120. Appl. Environ. Microbiol. 2009, 75, 7509–7518. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Dueñas, F.J.; Barrasa, J.M.; Sánchez-García, M.; Camarero, S.; Miyauchi, S.; Serrano, A.; Linde, D.; Babiker, R.; Drula, E.; Ayuso-Fernández, I.; et al. Genomic analysis enlightens Agaricales lifestyle evolution and increasing peroxidase diversity. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Sugano, Y. A structural and functional perspective of DyP-type peroxidase family. Arch. Biochem. Biophys. 2015, 574, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Linde, D.; Pogni, R.; Cañellas, M.; Lucas, F.; Guallar, V.; Baratto, M.C.; Sinicropi, A.; Sáez-Jiménez, V.; Coscolín, C.; Romero, A.; et al. Catalytic surface radical in dye-decolorizing peroxidase: A computational, spectroscopic and directed mutagenesis study. Biochem. J. 2015, 466, 253–262. [Google Scholar] [CrossRef]
- Acebes, S.; Ruiz-Dueñas, F.J.; Toubes, M.; Sáez-Jiménez, V.; Pérez-Boada, M.; Lucas, F.; Martínez, A.T.; Guallar, V. Mapping the long-range electron transfer route in ligninolytic peroxidases. J. Phys. Chem. B 2017, 121, 3946–3954. [Google Scholar] [CrossRef]
- Chao, C.; Tao, L. Bacterial dye-decolorizing peroxidases: Biochemical properties and biotechnological opportunities. Phys. Sci. Rev. 2016, 1, 20160051. [Google Scholar] [CrossRef] [Green Version]
- Goblirsch, B.; Kurker, R.C.; Streit, B.R.; Wilmot, C.M.; Dubois, J.L. Chlorite dismutases, DyPs, and EfeB: 3 microbial heme enzyme families comprise the CDE structural superfamily. J. Mol. Biol. 2011, 408, 379–398. [Google Scholar] [CrossRef] [Green Version]
- Rost, B. Protein structures sustain evolutionary drift. Fold. Des. 1997, 2, S19–S24. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Barros, T.; Chang, M.C.Y. Identification and characterization of a multifunctional dye peroxidase from a lignin-reactive bacterium. ACS Chem. Biol. 2012, 7, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shrestha, R.; Jia, K.; Gao, P.F.; Geisbrecht, B.V.; Bossmann, S.H.; Shi, J.; Li, P. Characterization of dye-decolorizing peroxidase (DyP) from Thermomonospora curvata reveals unique catalytic properties of A-type DyPs. J. Biol. Chem. 2015, 290, 23447–23463. [Google Scholar] [CrossRef] [Green Version]
- Liers, C.; Bobeth, C.; Pecyna, M.; Ullrich, R.; Hofrichter, M. DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl. Microbiol. Biotechnol. 2010, 85, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Dueñas, F.J.; Morales, M.; García, E.; Miki, Y.; Martínez, M.J.; Martínez, A.T. Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J. Exp. Bot. 2009, 60, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Boada, M.; Doyle, W.A.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Martínez, A.T.; Smith, A.T. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzyme Microb. Technol. 2002, 30, 518–524. [Google Scholar] [CrossRef]
- Doyle, W.A.; Smith, A.T. Expression of lignin peroxidase H8 in Escherichia coli: Folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem. J. 1996, 315, 15–19. [Google Scholar] [CrossRef] [Green Version]
- van Bloois, E.; Pazmino, D.E.T.; Winter, R.T.; Fraaije, M.W. A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl. Microbiol. Biotechnol. 2010, 86, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Habib, M.H.; Rozeboom, H.J.; Fraaije, M.W. Characterization of a new DyP-peroxidase from the alkaliphilic cellulomonad, Cellulomonas bogoriensis. Molecules 2019, 24, 1208. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Boada, M.; Ruiz-Dueñas, F.J.; Pogni, R.; Basosi, R.; Choinowski, T.; Martínez, M.J.; Piontek, K.; Martínez, A.T. Versatile peroxidase oxidation of high redox potential aromatic compounds: Site-directed mutagenesis, spectroscopic and crystallographic investigations of three long-range electron transfer pathways. J. Mol. Biol. 2005, 354, 385–402. [Google Scholar] [CrossRef]
- Morales, M.; Mate, M.J.; Romero, A.; Martínez, M.J.; Martínez, A.T.; Ruiz-Dueñas, F.J. Two oxidation sites for low redox-potential substrates: A directed mutagenesis, kinetic and crystallographic study on Pleurotus eryngii versatile peroxidase. J. Biol. Chem. 2012, 287, 41053–41067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso-Fernández, I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Experimental recreation of the evolution of lignin degrading enzymes from the Jurassic to date. Biotechnol. Biofuels 2017, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fueyo, E.; Davó-Siguero, I.; Almendral, D.; Linde, D.; Baratto, M.C.; Pogni, R.; Romero, A.; Guallar, V.; Martínez, A.T. Description of a non-canonical Mn(II)-oxidation site in peroxidases. ACS Catal. 2018, 8, 8386–8395. [Google Scholar] [CrossRef]
- Fernández-Fueyo, E.; Linde, D.; Almendral, D.; López-Lucendo, M.F.; Ruiz-Dueñas, F.J.; Martínez, A.T. Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). Appl. Microbiol. Biotechnol. 2015, 99, 8927–8942. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.N.; Singh, R.; Grigg, J.C.; Murphy, M.E.P.; Bugg, T.D.H.; Eltis, L.D. Characterization of dye-decolorizing peroxidases from Rhodococcus jostii RHA1. Biochemistry 2011, 50, 5108–5119. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.; Brissos, V.; Gabriel, A.; Catarino, T.; Turner, D.L.; Todorovic, S.; Martins, L.O. An integrated view of redox and catalytic properties of B-type PpDyP from Pseudomonas putida MET94 and its distal variants. Arch. Biochem. Biophys. 2015, 574, 99–107. [Google Scholar] [CrossRef]
- Rahmanpour, R.; Bugg, T.D. Characterisation of DyP-type peroxidases from Pseudomonas fluorescens Pf-5: Oxidation of Mn(II) and polymeric lignin by Dyp1B. Arch. Biochem. Biophys. 2015, 574, 93–98. [Google Scholar] [CrossRef]
- Rahmanpour, R.; Rea, D.; Jamshidi, S.; Fueloep, V.; Bugg, T.D. Structure of Thermobifida fusca DyP-type peroxidase and activity towards Kraft lignin and lignin model compounds. Arch. Biochem. Biophys. 2016, 594, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Loncar, N.; Colpa, D.I.; Fraaije, M.W. Exploring the biocatalytic potential of a DyP-type peroxidase by profiling the substrate acceptance of Thermobifida fusca DyP peroxidase. Tetrahedron 2016, 72, 7276–7281. [Google Scholar] [CrossRef]
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a bacterial DyP-type peroxidase for enhanced oxidation of lignin-related phenolics at alkaline pH. ACS Catal. 2017, 7, 3454–3465. [Google Scholar] [CrossRef]
- Ahmad, M.; Roberts, J.N.; Hardiman, E.M.; Singh, R.; Eltis, L.D.; Bugg, T.D.H. Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 2011, 50, 5096–5107. [Google Scholar] [CrossRef]
- Sezer, M.; Santos, A.; Kielb, P.; Pinto, T.; Martins, L.O.; Todorovic, S. Distinct structural and redox properties of the heme active site in bacterial dye decolorizing peroxidase-type peroxidases from two subfamilies: Resonance raman and electrochemical study. Biochemistry 2013, 52, 3074–3084. [Google Scholar] [CrossRef] [PubMed]
- Millis, C.D.; Cai, D.; Stankovich, M.T.; Tien, M. Oxidation-reduction potentials and ionization states of extracellular peroxidases from the lignin-degrading fungus Phanerochaete chrysosporium. Biochemistry 1989, 28, 8484–8489. [Google Scholar] [CrossRef] [PubMed]
- Ayala, M.; Roman, R.; Vázquez-Duhalt, R. A catalytic approach to estimate the redox potential of heme-peroxidases. Biochem. Biophys. Res. Commun. 2007, 357, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Ayuso-Fernández, I.; De Lacey, A.L.; Cañada, F.J.; Ruiz-Dueñas, F.J.; Martínez, A.T. Increase of redox potential during the evolution of enzymes degrading recalcitrant lignin. Chem. Eur. J. 2019, 25, 2708–2712. [Google Scholar] [CrossRef] [Green Version]
- Battistuzzi, G.; Bellei, M.; Bortolotti, C.A.; Sola, M. Redox properties of heme peroxidases. Arch. Biochem. Biophys. 2010, 500, 21–36. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Arnhold, J.; Jantschko, W.; Zederbauer, M.; Jakopitsch, C.; Obinger, C. Standard reduction potentials of all couples of the peroxidase cycle of lactoperoxidase. J. Inorg. Biochem. 2005, 99, 1220–1229. [Google Scholar] [CrossRef]
- Liers, C.; Aranda, E.; Strittmatter, E.; Piontek, K.; Plattner, D.A.; Zorn, H.; Ullrich, R.; Hofrichter, M. Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J. Mol. Catal. B Enzym. 2014, 103, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Tolbert, A.; Akinosho, H.; Khunsupat, R.; Naskar, A.K.; Ragauskas, A.J. Characterization and analysis of the molecular weight of lignin for biorefining studies. Biofuels Bioprod. Biorefining 2014, 8, 836–856. [Google Scholar] [CrossRef]
- Ayuso-Fernández, I.; Ruiz-Dueñas, F.J.; Martínez, A.T. Evolutionary convergence in lignin degrading enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 6428–6433. [Google Scholar] [CrossRef] [Green Version]
- Doyle, W.A.; Blodig, W.; Veitch, N.C.; Piontek, K.; Smith, A.T. Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry 1998, 37, 15097–15105. [Google Scholar] [CrossRef]
- Mester, T.; Ambert-Balay, K.; Ciofi-Baffoni, S.; Banci, L.; Jones, A.D.; Tien, M. Oxidation of a tetrameric nonphenolic lignin model compound by lignin peroxidase. J. Biol. Chem. 2001, 276, 22985–22990. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Jiménez, V.; Rencoret, J.; Rodríguez-Carvajal, M.A.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Role of surface tryptophan for peroxidase oxidation of nonphenolic lignin. Biotechnol. Biofuels 2016, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Jiménez, V.; Baratto, M.C.; Pogni, R.; Rencoret, J.; Gutiérrez, A.; Santos, J.I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Demonstration of lignin-to-peroxidase direct electron transfer. A transient-state kinetics, directed mutagenesis, EPR and NMR study. J. Biol. Chem. 2015, 290, 23201–23213. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Ichinose, H.; Wariishi, H. Determination of a catalytic tyrosine in Trametes cervina lignin peroxidase with chemical modification techniques. Biotechnol. Lett. 2011, 33, 1423–1427. [Google Scholar] [CrossRef]
- Miki, Y.; Pogni, R.; Acebes, S.; Lucas, F.; Fernández-Fueyo, E.; Baratto, M.C.; Fernández, M.I.; de los Ríos, V.; Ruiz-Dueñas, F.J.; Sinicropi, A.; et al. Formation of a tyrosine adduct involved in lignin degradation by Trametopsis cervina lignin peroxidase: A novel peroxidase activation mechanism. Biochem. J. 2013, 452, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, E.; Serrer, K.; Liers, C.; Ullrich, R.; Hofrichter, M.; Piontek, K.; Schleicher, E.; Plattner, D.A. The toolbox of Auricularia auricula-judae dye-decolorizing peroxidase. Identification of three new potential substrate-interaction sites. Arch. Biochem. Biophys. 2015, 574, 75–85. [Google Scholar] [CrossRef]
- Shrestha, R.; Chen, X.; Ramyar, K.X.; Hayati, Z.; Carlson, E.A.; Bossmann, S.H.; Song, L.; Geisbrecht, B.V.; Li, P. Identification of surface-exposed protein radicals and a substrate oxidation site in A-class dye-decolorizing peroxidase from Thermomonospora curvata. ACS Catal. 2016, 6, 8036–8047. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.; Huang, G.; Meekins, D.A.; Geisbrecht, B.V.; Li, P. Mechanistic insights into dye-decolorizing peroxidase revealed by solvent isotope and viscosity effects. ACS Catal. 2017, 7, 6352–6364. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yuan, Z.; Wang, J.; Cui, Y.; Liu, S.; Ma, Y.; Gu, L.; Xu, S. Crystal structure and biochemical features of dye-decolorizing peroxidase YfeX from Escherichia coli O157 Asp143 and Arg232 play divergent roles toward different substrates. Biochem. Biophys. Res. Commun. 2017, 484, 40–44. [Google Scholar] [CrossRef]
- Pfanzagl, V.; Bellei, M.; Hofbauer, S.; Laurent, C.V.F.P.; Furtmuller, P.G.; Oostenbrink, C.; Battistuzzi, G.; Obinger, C. Redox thermodynamics of B-class dye-decolorizing peroxidases. J. Inorg. Biochem. 2019, 199, 110761. [Google Scholar] [CrossRef]
- Rahmanpour, R.; Ehibhatiomhan, A.; Huang, Y.L.; Ashley, B.; Rashid, G.M.; Mendel-Williams, S.; Bugg, T.D.H. Protein engineering of Pseudomonas fluorescens peroxidase Dyp1B for oxidation of phenolic and polymeric lignin substrates. Enzyme Microb. Technol. 2019, 123, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Wang, X.P.; Cao, L.F.; Xu, M.Y. Lignin catabolic pathways reveal unique characteristics of dye-decolorizing peroxidases in Pseudomonas putida. Environ. Microbiol. 2019, 21, 1847–1863. [Google Scholar] [CrossRef]
- Santos, A.; Mendes, S.; Brissos, V.; Martins, L.O. New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: Towards biotechnological applications. Appl. Microbiol. Biotechnol. 2014, 98, 2053–2065. [Google Scholar] [CrossRef]
- Sezer, M.; Genebra, T.; Mendes, S.; Martins, L.O.; Todorovic, S. A DyP-type peroxidase at a bio-compatible interface: Structural and mechanistic insights. Soft Matter. 2012, 8, 10314–10321. [Google Scholar] [CrossRef]
- Uchida, T.; Sasaki, M.; Tanaka, Y.; Ishimorit, K. A dye-decolorizing peroxidase from Vibrio cholerae. Biochemistry 2015, 54, 6610–6621. [Google Scholar] [CrossRef]
- Sugawara, K.; Nishihashi, Y.; Narioka, T.; Yoshida, T.; Morita, M.; Sugano, Y. Characterization of a novel DyP-type peroxidase from Streptomyces avermitilis. J. Biosci. Bioeng. 2017, 123, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Salvachúa, D.; Prieto, A.; Martínez, A.T.; Martínez, M.J. Characterization of a novel dye-decolorizing peroxidase (DyP)-type enzyme from Irpex lacteus and its application in enzymatic hydrolysis of wheat straw. Appl. Environ. Microbiol. 2013, 79, 4316–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; Miki, Y.; Martínez, M.J.; Hammel, K.E.; Martínez, A.T. Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. J. Biol. Chem. 2012, 287, 16903–16916. [Google Scholar] [CrossRef] [Green Version]
- Ayuso-Fernández, I.; Rencoret, J.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Peroxidase evolution in white-rot fungi follows wood lignin evolution in plants. Proc. Natl. Acad. Sci. USA 2019, 116, 17900–17905. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Express. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Berry, E.A.; Trumpower, B.L. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 1987, 161, 1–15. [Google Scholar] [CrossRef]
- Moss, D.S.; Naberdryk, E.; Breton, J.; Mäntele, W. Redox-linked conformational changes in proteins detected by a combination of infrared spectroscopy and protein electrochemistry. Eur. J. Biochem. 1990, 187, 565–572. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ABTS | DMP | RB19 | Mn2+ | VA | H2O2 1 | ||
|---|---|---|---|---|---|---|---|
| AspDyP2 | Km | 165 ± 18 | 177 ± 34 | 20 ± 4 | 78 ± 18 | - | 42 ± 9 |
| kcat | 30.0 ± 0.9 | 94.0 ± 4.6 | 26.0 ± 2.0 | 42.0 ± 4.0 | 0 | 46.0 ± 4.6 | |
| kcat/Km | 180 ± 20 | 530 ± 80 | 1300 ± 200 | 540 ± 80 | - | 1100 ± 100 | |
| TcuDyP | Km | 309 ± 95 | 2 ± 0.3 | 7 ± 2 | - | - | 157 ± 29 |
| kcat | 53.0 ± 6.1 | 2.7 ± 0.4 | 1.3 ± 0.1 | 0 | 0 | 59.0 ± 1.2 | |
| kcat/Km | 170 ± 30 | 1300 ± 100 | 200 ± 40 | - | - | 380 ± 10 | |
| AauDyP 2 | Km | 123 ± 7 | 703 ± 60 | 90 ± 10 | - | - | 137 ± 26 |
| kcat | 225.0 ± 3.0 | 120.0 ± 3.0 | 224.0 ± 10.0 | 0 | 0 | 223.0 ± 23.0 | |
| kcat/Km | 1800 ± 90 | 173 ± 22 | 2400 ± 180 | - | 0.1 ± 0 | 1600 ± 200 | |
| PerVPL 3 | Km | 3 ± 0.2 | 78 ± 8 | 33 ± 8 | 181 ± 10 | 4130 ± 320 | 150 ± 16 |
| kcat | 8.1 ± 0.2 | 5.6 ± 0.1 | 8.5 ± 0.7 | 275.0 ± 4.0 | 9.5 ± 0.2 | 36 ± 1 | |
| kcat/Km | 2700 ± 140 | 71 ± 6 | 260 ± 60 | 1520 ± 70 | 2 ± 0.1 | 241 ± 17 | |
| PchLiPA 4 | Km | 21 ± 2 | 7 ± 1 | 34 ± 13 | - | 79 ± 18 | 82 ± 4 |
| kcat | 6.5 ± 0.2 | 4.0 ± 0.1 | 1.5 ± 0.3 | 0 | 16.2 ± 0.8 | 35 ± 1 | |
| kcat/Km | 300 ± 25 | 600 ± 36 | 45 ± 20 | - | 205 ± 4 | 420 ± 16 |
| Phenolic GGE | Nonphenolic VGE | |||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| AspDyP2 | 179 ± 39 | 12.0 ± 1.2 | 67.0 ± 8.5 | - | 0 | - |
| TcuDyP | 80 ± 16 | 1.1 ± 0.1 | 14.2 ± 1.8 | - | 0 | - |
| AauDyP | - | 0 | 19.8 ± 1.1 1 | 448 ± 59 | 0.3 ± 0.01 | 0.7 ± 0.1 |
| PerVPL | 206 ± 17 | 2.7 ± 0.1 | 13.2 ± 0.7 | 659 ± 80 | 1.2 ± 0.1 | 1.8 ± 0.1 |
| PchLiPA | 208 ± 41 | 1.3 ± 0.1 | 6.5 ± 0.7 | 88 ± 3 | 1.3 ± 0.2 | 14.7 ± 0.4 |
| Eo’(CI/RS) | Eo’(CI/CII) 1 | Eo’(CII/RS) 2 | |
|---|---|---|---|
| AspDyP2 | 1.423 ± 0.002 | 1.573 ± 0.009 | 1.273 ± 0.013 |
| TcuDyP | 1.371 ± 0.004 | 1.510 ± 0.002 | 1.232 ± 0.010 |
| AauDyP | 1.368 ± 0.004 | 1.465 ± 0.005 | 1.271 ± 0.003 |
| PerVPL | 1.383 ± 0.004 | 1.436 ± 0.003 | 1.330 ± 0.011 |
| PchLiPA | 1.402 ± 0.002 | 1.520 ± 0.006 | 1.283 ± 0.007 |
| Type | Organism | RB19 | DMP | VA | Mn2+ | Lignin Decay Assays | Fe3+/Fe2+ | CII/RS | PDB | Refs |
|---|---|---|---|---|---|---|---|---|---|---|
| DyP-I | Bacillus subtilis | + | nd | tr | nd | Good GGE (very low VGE) oxidation (HPLC) | −0.040 | nd | nd | [26,56] |
| DyP-I | Thermobifida fusca | + | + | tr | 0 | Guaiacol, kraft lignin, GGE (no VGE) oxidation | nd | nd | 5fw4 | [42,52,53] |
| DyP-I | Thermomonospora curvata | 2300–7800 · 103 | [1.3 · 103] | [0] | [0] | GGE [no VGE] oxidation [HPLC, kinetics] MeO-mandelic acid oxidation | [−0.136] | [1.232] | 5jxu | [37,72] [this study] |
| DyP-P | Enterobacter lignolyticus | 4200 · 103 | nd | 0 | tr | Low oxidation of phenols (no VA) but good dye oxidation | −0.290 | nd | 5vj0 | [73] |
| DyP-P | Escherichia coli | + | nd | nd | nd | Oxidation of phenols | nd | nd | 5gt2 | [74] |
| DyP-P | Klebsiella pneumoniae | nd | nd | nd | nd | nd | −0.350 | nd | 6fks | [75] |
| DyP-P | Pseudomonas fluorescens (DyP1B) | nd | 0.53 · 103 | nd | 0.33–0.66 · 103 | Kraft lignin (465 nm; with kcat/Km 140 · 103) wheat straw (HPLC) | nd | nd | nd | [51,76] |
| DyP-P | Pseudomonas putida | + | 0.7 · 103 | tr | 52 · 103 | Kraft lignin (LC-MS) GGE (HPLC) | −0.260 | nd | nd | [54,77,78,79] |
| DyP-P | Pseudomonas sp | nd | 9.3 · 103 | nd | 34 · 103 | Guaiacol oxidation, phenols from lignin | nd | nd | nd | [19] |
| DyP-P | Rhodococcus jostii (DyPB) | nd | nd | nd | 0.025 · 103 | Nitrated lignin assay, kraft lignin, GGE (HPLC, stopped flow) oxidation | nd | nd | 3veg | [49,55] |
| DyP-P | Vibrio cholerae | 26 · 103 | nd | nd | nd | Guaiacol oxidation | nd | nd | 5de0 | [80] |
| DyP-V | Anabaena sp | + | nd | nd | nd | Oxidation of phenols | nd | nd | 5c2i | [27] |
| DyP-V | Amycolatopsis sp (DyP2) | [1300 · 103] | [0.53 · 103] | [0] | [0.54]–120 · 103 | [GGE (no VGE) oxidation (HPLC, kinetics)], MeO-mandelic oxidation | [−0.085] | [1.273] | 4g2c | [36] [this study] |
| DyP-V | Streptomyces avermitilis | nd | 0.067 · 103 | nd | nd | Anthraquinonic dye oxidation | nd | nd | nd | [81] |
| DyP-V | Auricularia auricula-judae | 4800 · 103 | 3900–2000 · 103 | 0.2–0.11 · 103 | 0 | [GGE and VGE (low) (HPLC, kinetics)] other nonphenolic dimers (low) | [−0.160] | [1.271] | 4au9 4w7j | [24,38] [this study] |
| DyP-V | Irpex lacteus | 5900 · 103 | 970 · 103 | 0.83 · 103 | 0 | Increased digestibility of treated wheat straw | nd | nd | nd | [82] |
| DyP-V | Pleurotus ostreatus (DyP4) | 1860 · 103 | 2120 · 103 | 0 | 196 · 103 | Phenolic lignin oxidation given Mn-oxidizing activity | nd | nd | 6fsk | [48] |
| LiP | Phanerochaete chrysosporium (LiPA) | [0.045 · 103] | 1720 · 103 | 92 · 103 | 0 | Lignosulfonate transient-state kinetics and NMR [GGE and VGE (HPLC, kinetics)] | [−0.090] | [1.330] | 1qpa | [83,84] [this study] |
| VP | Pleurotus eryngii (VPL) | [0.26 · 103] | 71 · 103 | 2.3 · 103 | 1520 · 103 | Lignosulfonate transient-state kinetics and NMR [GGE and VGE (HPLC, kinetics)] | [−0.087] | [1.283] | 2boq | [67,68,83] [this study] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linde, D.; Ayuso-Fernández, I.; Laloux, M.; Aguiar-Cervera, J.E.; de Lacey, A.L.; Ruiz-Dueñas, F.J.; Martínez, A.T. Comparing Ligninolytic Capabilities of Bacterial and Fungal Dye-Decolorizing Peroxidases and Class-II Peroxidase-Catalases. Int. J. Mol. Sci. 2021, 22, 2629. https://doi.org/10.3390/ijms22052629
Linde D, Ayuso-Fernández I, Laloux M, Aguiar-Cervera JE, de Lacey AL, Ruiz-Dueñas FJ, Martínez AT. Comparing Ligninolytic Capabilities of Bacterial and Fungal Dye-Decolorizing Peroxidases and Class-II Peroxidase-Catalases. International Journal of Molecular Sciences. 2021; 22(5):2629. https://doi.org/10.3390/ijms22052629
Chicago/Turabian StyleLinde, Dolores, Iván Ayuso-Fernández, Marcos Laloux, José E. Aguiar-Cervera, Antonio L. de Lacey, Francisco J. Ruiz-Dueñas, and Angel T. Martínez. 2021. "Comparing Ligninolytic Capabilities of Bacterial and Fungal Dye-Decolorizing Peroxidases and Class-II Peroxidase-Catalases" International Journal of Molecular Sciences 22, no. 5: 2629. https://doi.org/10.3390/ijms22052629
APA StyleLinde, D., Ayuso-Fernández, I., Laloux, M., Aguiar-Cervera, J. E., de Lacey, A. L., Ruiz-Dueñas, F. J., & Martínez, A. T. (2021). Comparing Ligninolytic Capabilities of Bacterial and Fungal Dye-Decolorizing Peroxidases and Class-II Peroxidase-Catalases. International Journal of Molecular Sciences, 22(5), 2629. https://doi.org/10.3390/ijms22052629