
Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins




Abstract
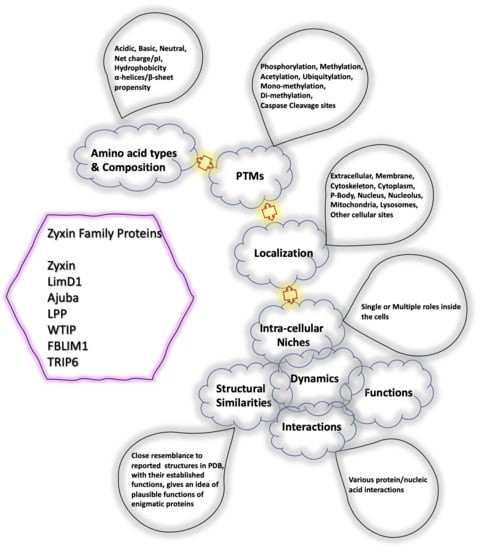
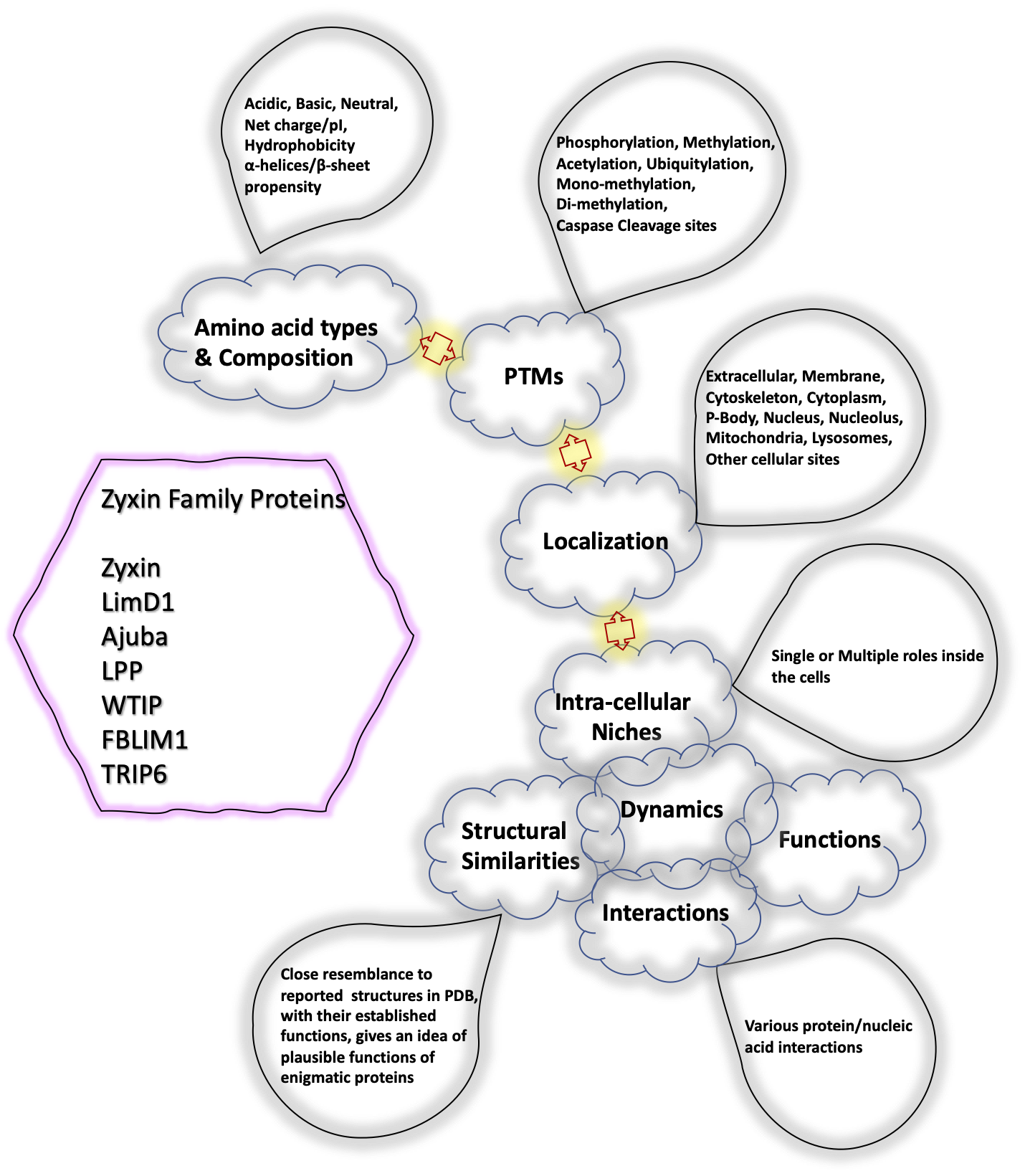
:
1. Introduction
2. Results
2.1. Human Zyxin Family Proteins Exhibit Variations in the Proline-Rich Region and Lim Edges
2.2. Impact of Post-Translational Modifications (PTMs) on the Isoelectric Point (pI) of Zyxin Family Proteins
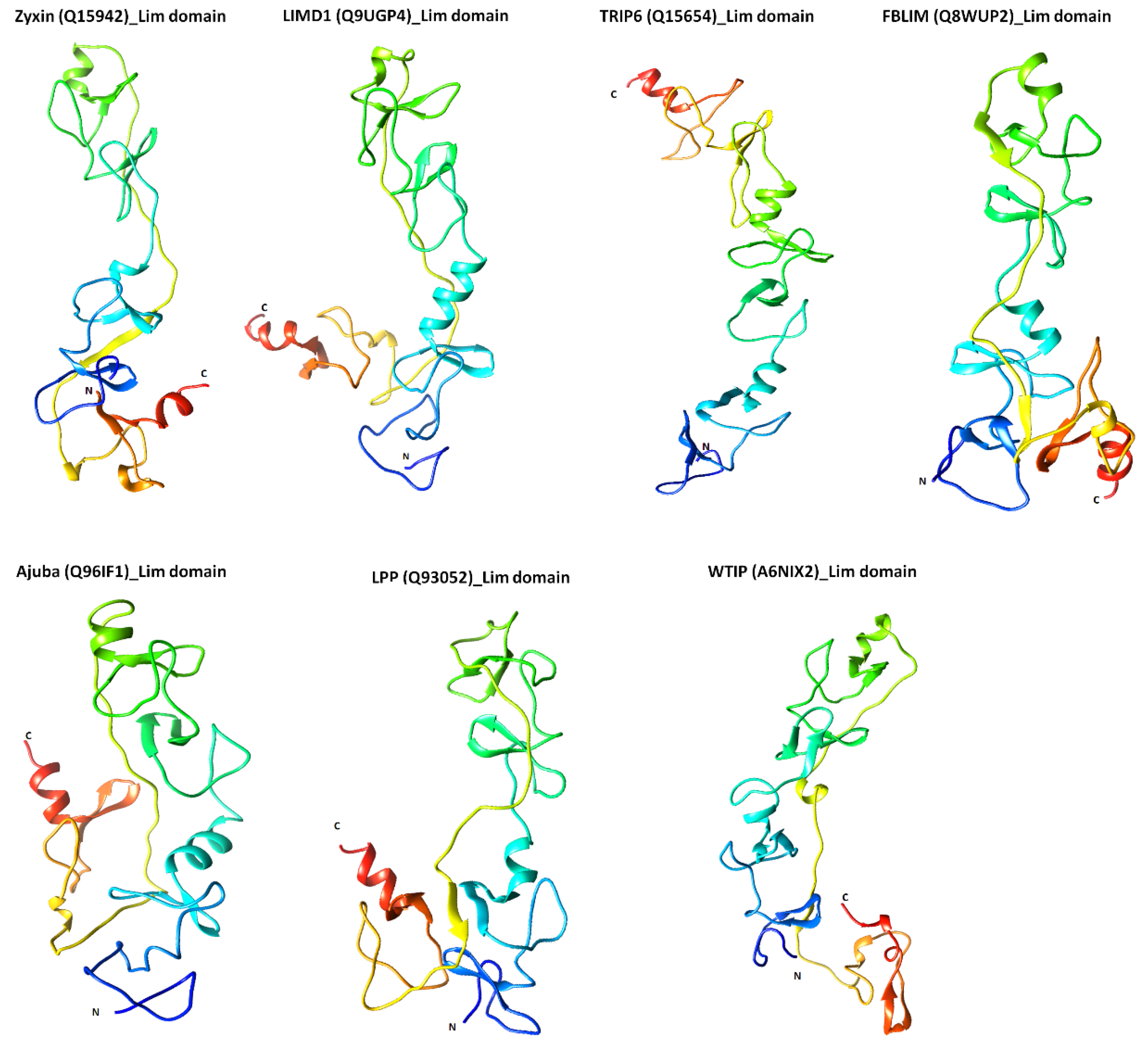
2.3. Structural Features of Zyxin Family Lim Domains Indicate Their Similarities with Transcription Factors
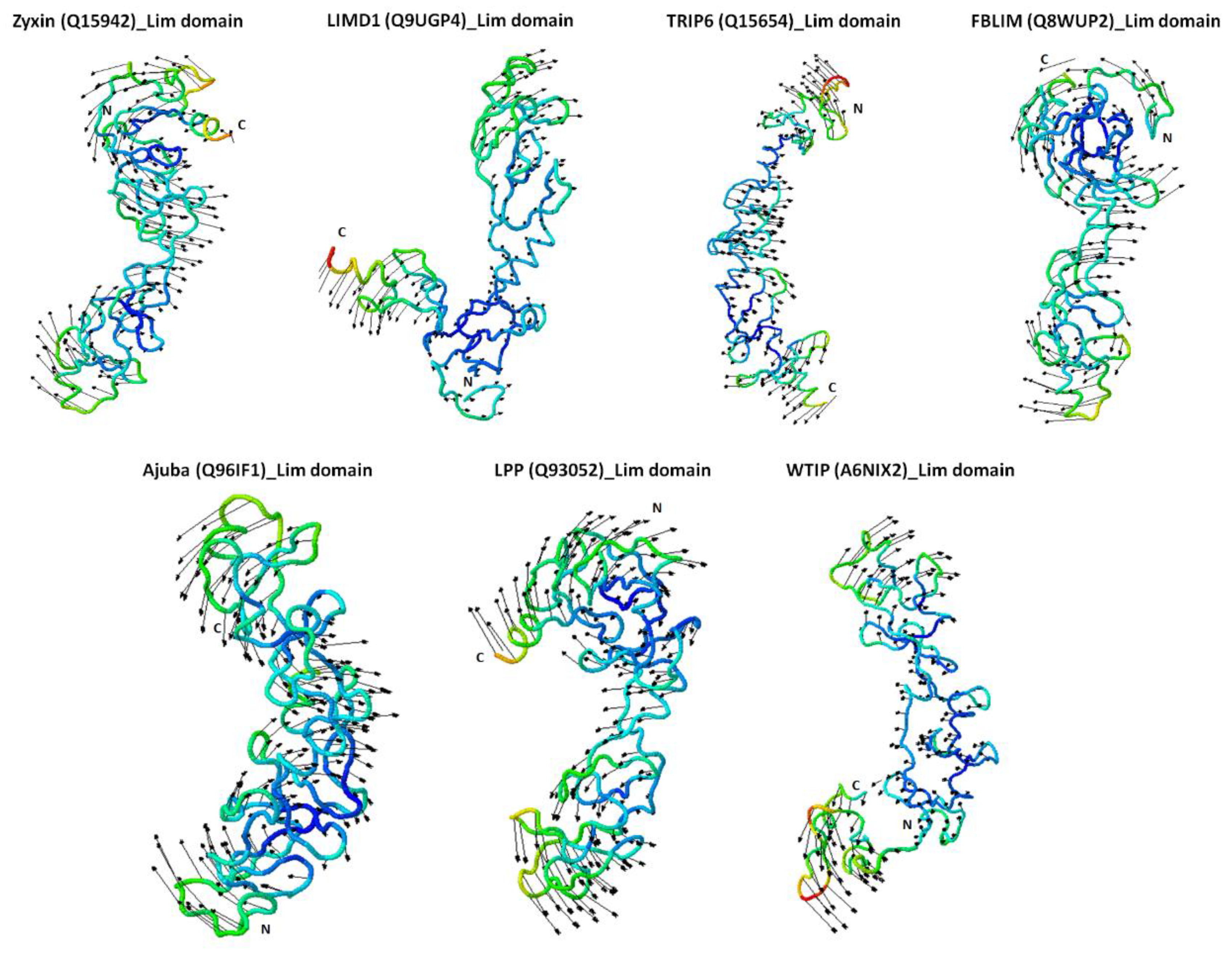
2.4. Zyxin Family Lim Domains Display Contrasting Dynamics with Large Inter-Domain Motion of Individual Lim1/2/3 Domains
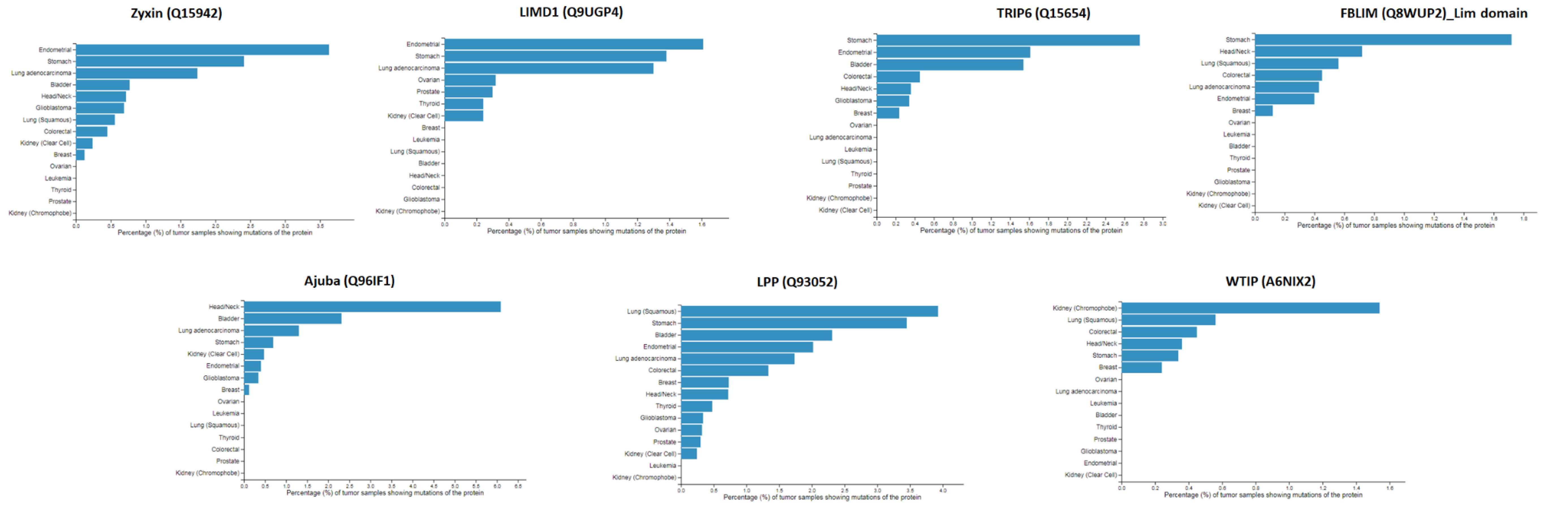
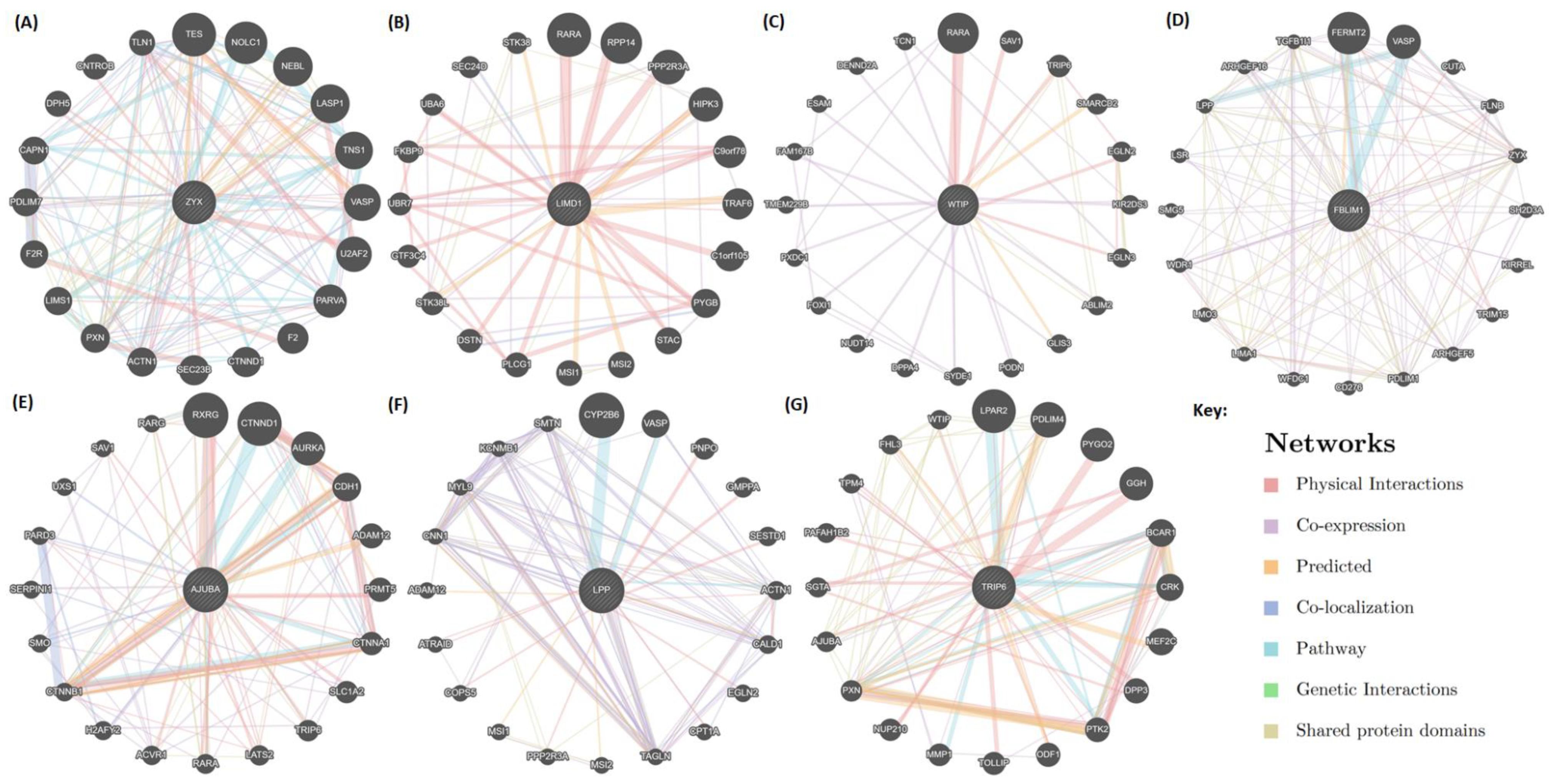
2.5. Mapping of Protein-Protein Interactions of the Zyxin Family Proteins Provides Insights into Their Crucial Role in Cancer Progression
3. Discussion
4. Materials and Methods
4.1. Sequence, Amino Acid Composition, Phylogeny, and Basic Biochemical Analysis
4.2. Structure Modeling, Validation and Alignment of Zyxin Family Lim Domains
Normal Mode Analysis (NMA), Elastic Network Modeling and Covariance Analysis
4.3. Protein-Protein Interactions of Zyxin Family Proteins
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kotb, A.; Hyndman, M.E.; Patel, T.R. The role of zyxin in regulation of malignancies. Heliyon 2018, 4, e00695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Gaag, E.J.; Leccia, M.T.; Dekker, S.K.; Jalbert, N.L.; Amodeo, D.M.; Byers, H.R. Role of zyxin in differential cell spreading and proliferation of melanoma cells and melanocytes. J. Investig. Dermatol. 2002, 118, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zeng, Y.; Cui, L.; Chen, X.; Stauffer, S.; Wang, Z.; Yu, F.; Lele, S.M.; Talmon, G.A.; Black, A.R.; et al. Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc. Natl. Acad. Sci. USA 2018, 115, E6760–E6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, P.; Holder, M.V.; Aerne, B.L.; Janody, F.; Tapon, N. Zyxin Antagonizes the FERM Protein Expanded to Couple F-Actin and Yorkie-Dependent Organ Growth. Curr. Biol. 2015, 25, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauskolb, C.; Pan, G.; Reddy, B.V.V.G.; Oh, H.; Irvine, K.D. Zyxin Links Fat Signaling to the Hippo Pathway. PLoS Biol. 2011, 9, e1000624. [Google Scholar] [CrossRef] [Green Version]
- Sy, S.M.-H.; Lai, P.B.-S.; Pang, E.; Wong, N.L.-Y.; To, K.-F.; Johnson, P.J.; Wong, N. Novel identification of zyxin upregulations in the motile phenotype of hepatocellular carcinoma. Mod. Pathol. 2006, 19, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-beta (TGF-beta)/Smad3 target gene that regulates lung cancer cell motility via integrin alpha5beta1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Yu, J.; Li, D.; Jiang, K.; Tang, Y.; Yang, M.; Shen, H.; Fang, X.; Ding, K.; Zheng, S.; et al. Zyxin as a potential cancer prognostic marker promotes the proliferation and metastasis of colorectal cancer cells. J. Cell. Physiol. 2019, 234, 15775–15789. [Google Scholar] [CrossRef] [PubMed]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.-M.; Luo, T.; Jiang, Y.; Wang, L.-H.; Luo, Y.; Chen, Q.; Yang, K.; Yuan, Y.; Luo, C.; Zhang, X.; et al. Zyxin (ZYX) promotes invasion and acts as a biomarker for aggressive phenotypes of human glioblastoma multiforme. Lab. Investig. 2020, 100, 812–823. [Google Scholar] [CrossRef]
- Wu, Z.; Qiu, M.; Mi, Z.; Meng, M.; Guo, Y.; Jiang, X.; Fang, J.; Wang, H.; Zhao, J.; Liu, Z.; et al. WT1-interacting protein inhibits cell proliferation and tumorigenicity in non-small-cell lung cancer via the AKT/FOXO1 axis. Mol. Oncol. 2019, 13, 1059–1074. [Google Scholar] [CrossRef] [Green Version]
- James, V.; Zhang, Y.; Foxler, D.E.; de Moor, C.H.; Kong, Y.W.; Webb, T.M.; Self, T.J.; Feng, Y.; Lagos, D.; Chu, C.Y.; et al. LIM-domain proteins, LIMD1, Ajuba, and WTIP are required for microRNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12499–12504. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Ma, L.; Dong, L.; Ma, L.; Zhao, Z.; Ma, L.; Zhou, W.; Fan, J.; Wu, C.; Yu, C.; et al. Migfilin protein promotes migration and invasion in human glioma through epidermal growth factor receptor-mediated phospholipase C-gamma and STAT3 protein signaling pathways. J. Biol. Chem. 2012, 287, 32394–32405. [Google Scholar] [CrossRef] [Green Version]
- Kiepas, A.; Voorand, E.; Senecal, J.; Ahn, R.; Annis, M.G.; Jacquet, K.; Tali, G.; Bisson, N.; Ursini-Siegel, J.; Siegel, P.M.; et al. The SHCA adapter protein cooperates with lipoma-preferred partner in the regulation of adhesion dynamics and invadopodia formation. J. Biol. Chem. 2020, 295, 10535–10559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyama, S.; Yoshida, M.; Yano, S.; Aiba, N.; Kohno, T.; Minamiya, Y.; Goto, A.; Tanaka, M. LPP inhibits collective cell migration during lung cancer dissemination. Oncogene 2016, 35, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, R.; Wang, X.; Song, D.; Cooke, N.E.; Liebhaber, S.A. The LIM/double zinc-finger motif functions as a protein dimerization domain. Proc. Natl. Acad. Sci. USA 1994, 91, 10655–10659. [Google Scholar] [CrossRef] [Green Version]
- Schmeichel, K.L.; Beckerle, M.C. The LIM domain is a modular protein-binding interface. Cell 1994, 79, 211–219. [Google Scholar] [CrossRef]
- Kadrmas, J.L.; Beckerle, M.C. The LIM domain: From the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 2004, 5, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, J.W.; Schmeichel, K.L.; Beckerle, M.C.; Winge, D.R. The LIM motif defines a specific zinc-binding protein domain. Proc. Natl. Acad. Sci. USA 1993, 90, 4404–4408. [Google Scholar] [CrossRef] [Green Version]
- Minor, D.L., Jr.; Kim, P.S. Measurement of the beta-sheet-forming propensities of amino acids. Nature 1994, 367, 660–663. [Google Scholar] [CrossRef]
- Singh, P.P.; Isambert, H. OHNOLOGS v2: A comprehensive resource for the genes retained from whole genome duplication in vertebrates. Nucleic Acids Res. 2020, 48, D724–D730. [Google Scholar] [CrossRef]
- La Cour, T.; Kiemer, L.; Mølgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Kiraga, J.; Mackiewicz, P.; Mackiewicz, D.; Kowalczuk, M.; Biecek, P.; Polak, N.; Smolarczyk, K.; Dudek, M.R.; Cebrat, S. The relationships between the isoelectric point and: Length of proteins, taxonomy and ecology of organisms. BMC Genom. 2007, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.; Ting, C.S.; King, J. Whole Proteome pI Values Correlate with Subcellular Localizations of Proteins for Organisms within the Three Domains of Life. Genome Res. 2001, 11, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Zhao, J.; Lubman, D.M.; Miller, F.R.; Barder, T.J. Protein pI shifts due to posttranslational modifications in the separation and characterization of proteins. Anal. Chem. 2005, 77, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus(R): Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.M.R.; Fradelizi, J.; Golsteyn, R.M.; Ayoubi, T.A.; Menichi, B.; Louvard, D.; Van De Ven, W.J.M.; Friederich, E. LPP, an Actin Cytoskeleton Protein Related to Zyxin, Harbors a Nuclear Export Signal and Transcriptional Activation Capacity. Mol. Biol. Cell 2000, 11, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P.S. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8, 463. [Google Scholar] [CrossRef] [Green Version]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Sam, V.; Tai, C.-H.; Garnier, J.; Gibrat, J.-F.; Lee, B.; Munson, P.J. ROC and confusion analysis of structure comparison methods identify the main causes of divergence from manual protein classification. BMC Bioinform. 2006, 7, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, J.E.; Ryan, D.P.; Sunde, M.; Maher, M.J.; Guss, J.M.; Visvader, J.E.; Matthews, J.M. Tandem LIM domains provide synergistic binding in the LMO4:Ldb1 complex. EMBO J. 2004, 23, 3589–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffries, C.M.; Graham, S.C.; Stokes, P.H.; Collyer, C.A.; Guss, J.M.; Matthews, J.M. Stabilization of a binary protein complex by intein-mediated cyclization. Protein Sci. 2006, 15, 2612–2618. [Google Scholar] [CrossRef] [Green Version]
- Bhati, M.; Lee, C.; Nancarrow, A.L.; Lee, M.; Craig, V.J.; Bach, I.; Guss, J.M.; Mackay, J.P.; Matthews, J.M. Implementing the LIM code: The structural basis for cell type-specific assembly of LIM-homeodomain complexes. EMBO J. 2008, 27, 2018–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, P.H.; Robertson, N.O.; Silva, A.P.G.; Estephan, T.; Trewhella, J.; Guss, J.M.; Matthews, J.M. Mutation in a flexible linker modulates binding affinity for modular complexes. Proteins 2019, 87, 425–429. [Google Scholar] [CrossRef]
- Gadd, M.S.; Jacques, D.A.; Nisevic, I.; Craig, V.J.; Kwan, A.H.; Guss, J.M.; Matthews, J.M. A Structural Basis for the Regulation of the LIM-Homeodomain Protein Islet 1 (Isl1) by Intra- and Intermolecular Interactions. J. Biol. Chem. 2013, 288, 21924–21935. [Google Scholar] [CrossRef] [Green Version]
- Racevskis, J.; Dill, A.; Sparano, J.A.; Ruan, H. Molecular cloning of LMO41, a new human LIM domain gene. Biochim. Biophys. Acta 1999, 1445, 148–153. [Google Scholar] [CrossRef]
- Vu, D.; Marin, P.; Walzer, C.; Cathieni, M.M.; Bianchi, E.N.; Saïdji, F.; Leuba, G.; Bouras, C.; Savioz, A. Transcription regulator LMO4 interferes with neuritogenesis in human SH-SY5Y neuroblastoma cells. Mol. Brain Res. 2003, 115, 93–103. [Google Scholar] [CrossRef]
- Sum, E.Y.M.; Peng, B.; Yu, X.; Chen, J.; Byrne, J.; Lindeman, G.J.; Visvader, J.E. The LIM Domain Protein LMO4 Interacts with the Cofactor CtIP and the Tumor Suppressor BRCA1 and Inhibits BRCA1 Activity. J. Biol. Chem. 2002, 277, 7849–7856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Schweppe, D.K.; Huttlin, E.L.; Harper, J.W.; Gygi, S.P. BioPlex Display: An Interactive Suite for Large-Scale AP–MS Protein–Protein Interaction Data. J. Proteome Res. 2018, 17, 722–726. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Licata, L.; Surdo, P.L.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-K.; Pai, C.-Y.; Huang, J.-Y.; Yeh, N.-H. Human Nopp140, Which Interacts with RNA Polymerase I: Implications for rRNA Gene Transcription and Nucleolar Structural Organization. Mol. Cell. Biol. 1999, 19, 8536–8546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.-B.; Liu, X.; Tang, X.; Fu, H.; Ye, K. Akt phosphorylation of zyxin mediates its interaction with acinus-S and prevents acinus-triggered chromatin condensation. Cell Death Differ. 2007, 14, 1688–1699. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, Y.Y.; Silverstein, S. Interaction of Zyxin, a Focal Adhesion Protein, with the E6 Protein from Human Papillomavirus Type 6 Results in Its Nuclear Translocation. J. Virol. 2001, 75, 11791–11802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkelman, M.; Balcıoğlu, H.E.; Klip, J.E.; Yan, K.; Verbeek, F.J.; Danen, E.H.J.; Van De Water, B. Cellular adhesome screen identifies critical modulators of focal adhesion dynamics, cellular traction forces and cell migration behaviour. Sci. Rep. 2016, 6, 31707. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Y.A.; Rehn, M.; Ekman, N.; Alitalo, K.; Vuori, K. p130Cas Couples the Tyrosine Kinase Bmx/Etk with Regulation of the Actin Cytoskeleton and Cell Migration. J. Biol. Chem. 2003, 278, 35636–35643. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, A.; Ohba, Y.; Kurokawa, K.; Matsuda, M.; Hattori, S. Novel function of Chat in controlling cell adhesion via Cas-Crk-C3G-pathway-mediated Rap1 activation. J. Cell Sci. 2002, 115 Pt 24, 4915–4924. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, A.J.; Allen, T.W. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim. Biophys. Acta 2013, 1828, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, U.; Chakrabarti, P. Assessing the role of tryptophan residues in the binding site. Protein Eng. 2001, 14, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Nielsen, J.E.; Shields, D.C.; Khaldi, N. Evolution of the isoelectric point of mammalian proteins as a consequence of indels and adaptive evolution. Proteins 2011, 79, 1635–1648. [Google Scholar] [CrossRef]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA 2010, 107, 9270–9274. [Google Scholar] [CrossRef] [Green Version]
- Koch, B.J.; Ryan, J.F.; Baxevanis, A.D. The Diversification of the LIM Superclass at the Base of the Metazoa Increased Subcellular Complexity and Promoted Multicellular Specialization. PLoS ONE 2012, 7, e33261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyanathan, K.; Peng, H.; Hou, Z.; Fredericks, W.J.; Goyal, R.K.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J. The Ajuba LIM Domain Protein Is a Corepressor for SNAG Domain–Mediated Repression and Participates in Nucleocytoplasmic Shuttling. Cancer Res. 2007, 67, 9097–9106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Kim, E.-J.; Hitomi, M.; Oh, S.-Y.; Jin, X.; Jeon, H.-M.; Beck, S.; Kim, J.-K.; Park, C.G.; Chang, S.-Y.; et al. The LIM-only transcription factor LMO2 determines tumorigenic and angiogenic traits in glioma stem cells. Cell Death Differ. 2015, 22, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Marie, H.; Pratt, S.J.; Betson, M.; Epple, H.; Kittler, J.T.; Meek, L.; Moss, S.J.; Troyanovsky, S.M.; Attwell, D.; Longmore, G.D.; et al. The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J. Biol. Chem. 2003, 278, 1220–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Garcia, I.; Rabbitts, T.H. The LIM domain: A new structural motif found in zinc-finger-like proteins. Trends Genet. 1994, 10, 315–320. [Google Scholar] [CrossRef]
- Wu, R.-Y.; Durick, K.; Songyang, Z.; Cantley, L.C.; Taylor, S.S.; Gill, G.N. Specificity of LIM Domain Interactions with Receptor Tyrosine Kinases. J. Biol. Chem. 1996, 271, 15934–15941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parshina, E.A.; Eroshkin, F.M.; Orlov, E.E.; Gyoeva, F.K.; Shokhina, A.G.; Staroverov, D.B.; Belousov, V.V.; Zhigalova, N.A.; Prokhortchouk, E.B.; Zaraisky, A.G.; et al. Cytoskeletal Protein Zyxin Inhibits the Activity of Genes Responsible for Embryonic Stem Cell Status. Cell Rep. 2020, 33, 108396. [Google Scholar] [CrossRef] [PubMed]
- Martynova, N.; Parshina, E.; Ermolina, L.; Zaraisky, A. The cytoskeletal protein Zyxin interacts with the zinc-finger transcription factor Zic1 and plays the role of a scaffold for Gli1 and Zic1 interactions during early development of Xenopus laevis. Biochem. Biophys. Res. Commun. 2018, 504, 251–256. [Google Scholar] [CrossRef]
- Kamberaj, H.; van der Vaart, A. Extracting the causality of correlated motions from molecular dynamics simulations. Biophys. J. 2009, 97, 1747–1755. [Google Scholar] [CrossRef] [Green Version]
- Goodey, N.M.; Benkovic, S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008, 4, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hammes, G.G. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry 2002, 41, 8221–8228. [Google Scholar] [CrossRef] [PubMed]
- Jarymowycz, V.A.; Stone, M.J. Fast Time Scale Dynamics of Protein Backbones: NMR Relaxation Methods, Applications, and Functional Consequences. Chem. Rev. 2006, 106, 1624–1671. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M.; UniProt, C. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Prot. Bioinfor. 2002, 1, 1–22. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Ba, A.N.N.; Pogoutse, A.; Provart, N.; Moses, A.M. NLStradamus: A simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinform. 2009, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Bordoli, L.; Schwede, T. Automated Protein Structure Modeling with SWISS-MODEL Workspace and the Protein Model Portal. Methods Mol. Biol. 2012, 857, 107–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- López-Blanco, J.R.; Reyes, R.; Aliaga, J.I.; Badia, R.M.; Chacon, P.; Quintana-Ortí, E.S. Exploring large macromolecular functional motions on clusters of multicore processors. J. Comput. Phys. 2013, 246, 275–288. [Google Scholar] [CrossRef]
- Nguyen, D.-T.; Mathias, S.L.; Bologa, C.; Brunak, S.; Fernandez, N.F.; Gaulton, A.; Hersey, A.; Holmes, J.; Jensen, L.J.; Karlsson, A.; et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017, 45, D995–D1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, D.T.J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A Reference Expression Database for the Meta-Analysis of Transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein (UNIPROT ID) | Cellular Role * | Cellular Localization | Isoelectric Point 1 | Amino Acids/ Molecular Weight (kDa) 1 | ||
|---|---|---|---|---|---|---|
| Full Protein | LIM DoMain | Full Protein | LIM Domain | |||
| Zyxin (Q15942) | Adhesion plague protein, signal transducer | Cytoskeleton, cytosol and nucleus | 6.22 | 7.16 | 572/61.27 | 189/21.10 |
| LimD1 (Q9UGP4) | Cell fate determination and a tumour suppressor | Predominantly in nucleus, P- body | 6.20 | 5.90 | 676/72.19 | 195/21.87 |
| Ajuba (Q96IF1) | Cell fate determination and repression of gene transcription | Cell membrane, cytoskeleton, centrosome, nucleus, P- body | 6.86 | 5.81 | 538/56.93 | 195/21.87 |
| LPP (Q93052) | Maintains cell shape, motility, and activation of gene transcription | Cell membrane and nucleus | 7.18 | 6.78 | 612/65.74 | 190/21.15 |
| WTIP (A6NIX2) | Cell fate determination, repression of gene transcription, and miRNA-mediated gene silencing | Nucleus, P- body | 8.53 | 7.50 | 430/45.12 | 195/21.80 |
| FBLIM1 (Q8WUP2) | Cell-ECM adhesion proteins and filamin-containing actin filaments | Focal adhesion sites | 5.71 | 7.79 | 373/40.66 | 190/21.70 |
| TRIP6 (Q15654) | Relays signals from the cell surface to the nucleus | Focal adhesion sites, cytoplasm, nucleus | 7.19 | 6.06 | 476/50.28 | 188/20.49 |
| Protein (Uniprot ID) | Percentage of PRR vs. Full Length * | Percentage of Lim Domain vs. Full Length * |
|---|---|---|
| Zyxin (Q15942) | 66.9% | 33.1% |
| LimD1 (Q9UGP4) | 69.3% | 30.7% |
| Ajuba (Q96IF1) | 62.8% | 37.2% |
| LPP (Q93052) | 67.4% | 32.6% |
| WTIP (A6NIX2) | 51.6% | 48.4% |
| FBLIM1 (Q8WUP2) | 48.2% | 51.8% |
| TRIP6 (Q15654) | 58.4% | 41.6% |
| Protein | pI | Number of Sites for PTM | ||||||
|---|---|---|---|---|---|---|---|---|
| Native 1 | Post PTM *,2 | Phosphorylation | Acety- Lation * | Ubiquity- Lation * | Monomethy- Lation * | Dimethy- Lation * | Caspase Cleavage Sites * | |
| Zyxin | 6.22 | 6.12–3.17 | 66 | 5 | 4 | 3 | 2 | 1 |
| LimD1 | 6.20 | 6.12–3.85 | 65 | - | 8 | 4 | - | - |
| Ajuba | 6.86 | 6.63–4.63 | 30 | - | 2 | 1 | - | - |
| LPP | 7.18 | 6.89–2.84 | 83 | 3 | 5 | 1 | 1 | - |
| WTIP | 8.53 | 8.36–6.22 | 11 | - | - | - | - | - |
| FBLIM1 | 5.71 | 5.60–4.96 | 9 | 1 | - | - | - | - |
| TRIP6 | 7.19 | 6.89–3.61 | 56 | - | - | 22 | 4 | - |
| Protein | SVM Score * | Prediction | Job Number on Webserver |
|---|---|---|---|
| Zyxin | 1.2919473 | DNA binding protein | 1710 |
| LimD1 | 0.90588128 | DNA binding protein | 4069 |
| Ajuba | 1.4081202 | DNA binding protein | 7780 |
| LPP | 0.11482763 | DNA binding protein | 1668 |
| WTIP | 1.7520286 | DNA binding protein | 1032 |
| FBLIM1 | 0.39872591 | DNA binding protein | 1215 |
| TRIP6 | 0.83398047 | DNA binding protein | 3994 |
| LIM Domains | Description of Structural Similarities Identified by DALI Server | |
|---|---|---|
| Unique | Common | |
| Zyxin | Lim domain-bin, Lim domain-BI, CRP1, Four and Half Lim domain protein 2, Rhombotin-2 | Fusion protein of LMO4 protein, Lim/Homeobox protein LHX4, Fusion protein of Lim domain transcription factor Fusion of Lim/Homeobox protein LHX3 Insulin gene enhancer protein ISL-1 Insulin gene enhancer P Fusion protein of Lim domain transcription factor Actin Like protein 7A |
| LimD1 | Lim domain-BI, Lim/Homeobox protein LHX4, Rhombotin-2, CRP1 | |
| Ajuba | Thyroid receptor-interacting protein 6, T-cell acute lymphocytic leukemia protein 1 | |
| LPP | Lim domain-BI, Rhombotin-2, Thyroid receptor-interacting protein 6 | |
| WTIP | Lim domain-bin, Lim domain-BI, Four and Half Lim domain protein 2, Rhombotin-2 | |
| FBLIM1 | Lim domain-BI, Four and Half Lim domain protein 2, Rhombotin-2, Thyroid receptor-interacting protein 6 | |
| TRIP6 | Rhombotin-2, T-cell acute lymphocytic leukemia protein 1, Four and Half Lim domains 2, Thyroid receptor-interacting protein 6, Rhombotin-2, Paxillin, PINCH, Lim domain-BI | |
| Gene A | Gene B * | Interaction Type | Direction Method |
|---|---|---|---|
| BVLF1 (Epstein-Barr Virus) | Zyx | Association | Tandem Affinity Purification |
| E6 (Human papillomavirus type 18) | Zyx | Association | Tandem Affinity Purification |
| E6 (Human papillomavirus type 6b) | Zyx | Physical Association | Anti-Tag Co-immunoprecipitation |
| E6 (Human papillomavirus type 6b) | Zyx | Direct Interaction | Pull Down |
| E6 (Human papillomavirus type 6b) | Zyx | Physical Association | Two Hybrid |
| HIPK3 (Homo sapiens) | Zyx | Physical Association | Two Hybrid |
| JUN (Homo sapiens) | Zyx | Association | Anti-Bait Co-immunoprecipitation |
| HIPK3 (Homo sapiens) | Zyx | Physical Association | Two Hybrid |
| JUN (Homo sapiens) | Zyx | Association | Anti-Bait Co-immunoprecipitation |
| NEK4 (Homo sapiens) | Zyx | Association | Anti-Tag Co-immunoprecipitation |
| NEK4 (Homo sapiens) | Zyx | Association | Anti-Tag Co-immunoprecipitation |
| NS1 (Influenza A virus) | Zyx | Physical Association | Two Hybrid Array |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddiqui, M.Q.; Badmalia, M.D.; Patel, T.R. Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins. Int. J. Mol. Sci. 2021, 22, 2647. https://doi.org/10.3390/ijms22052647
Siddiqui MQ, Badmalia MD, Patel TR. Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins. International Journal of Molecular Sciences. 2021; 22(5):2647. https://doi.org/10.3390/ijms22052647
Chicago/Turabian StyleSiddiqui, M. Quadir, Maulik D. Badmalia, and Trushar R. Patel. 2021. "Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins" International Journal of Molecular Sciences 22, no. 5: 2647. https://doi.org/10.3390/ijms22052647
APA StyleSiddiqui, M. Q., Badmalia, M. D., & Patel, T. R. (2021). Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins. International Journal of Molecular Sciences, 22(5), 2647. https://doi.org/10.3390/ijms22052647