
Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization


Abstract
:1. Introduction
2. Results and Discussion
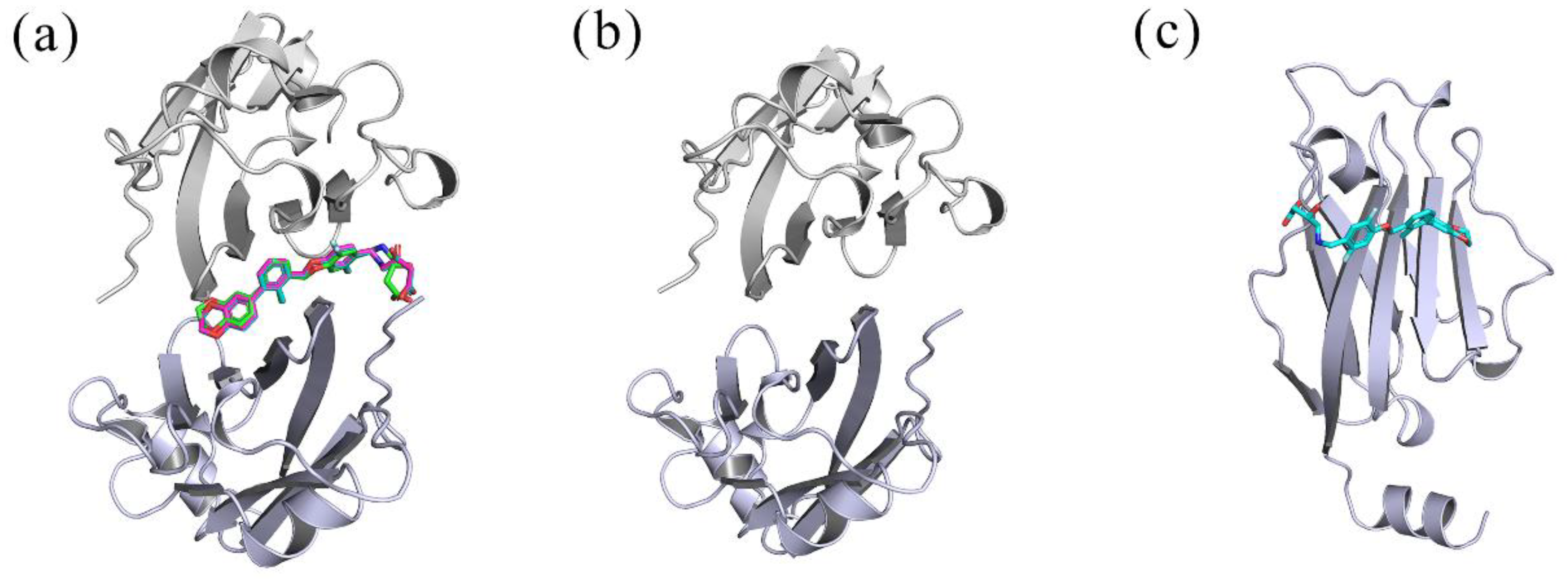
2.1. Docking
2.2. RMSD
2.3. RMSF
2.4. Binding Free Energy
2.5. Per-Residue Energy Decomposition
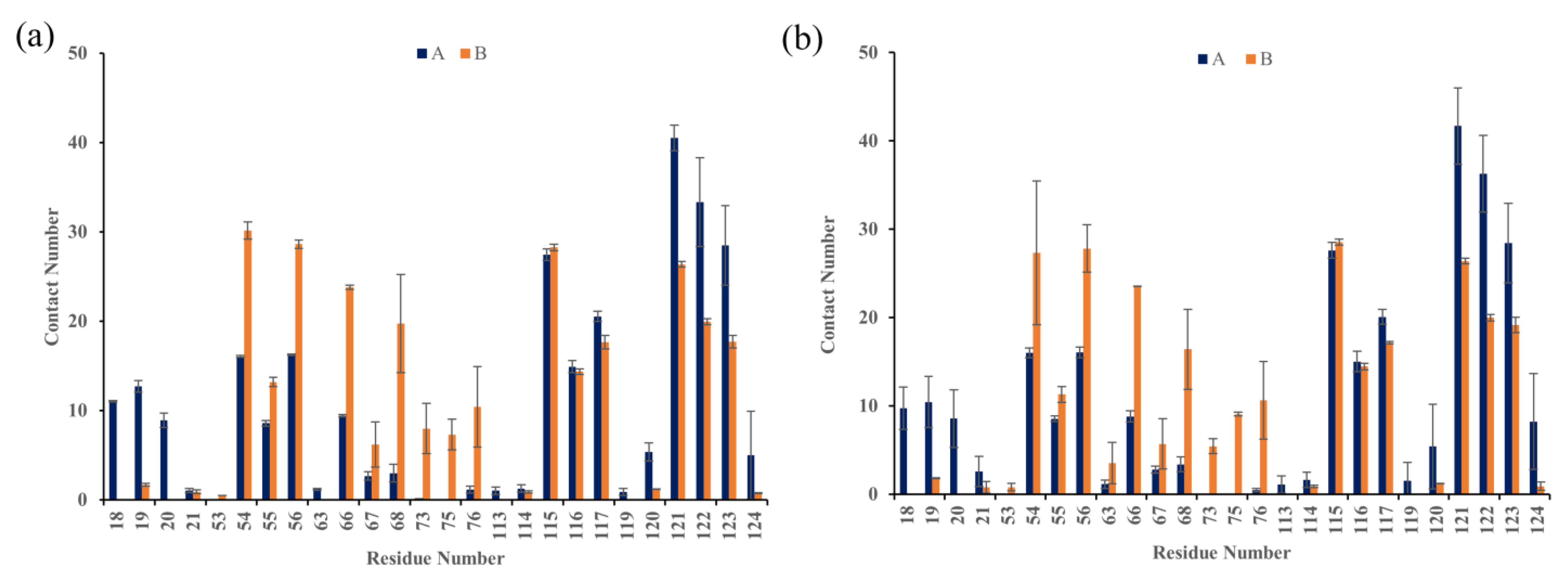
2.6. Contact Numbers
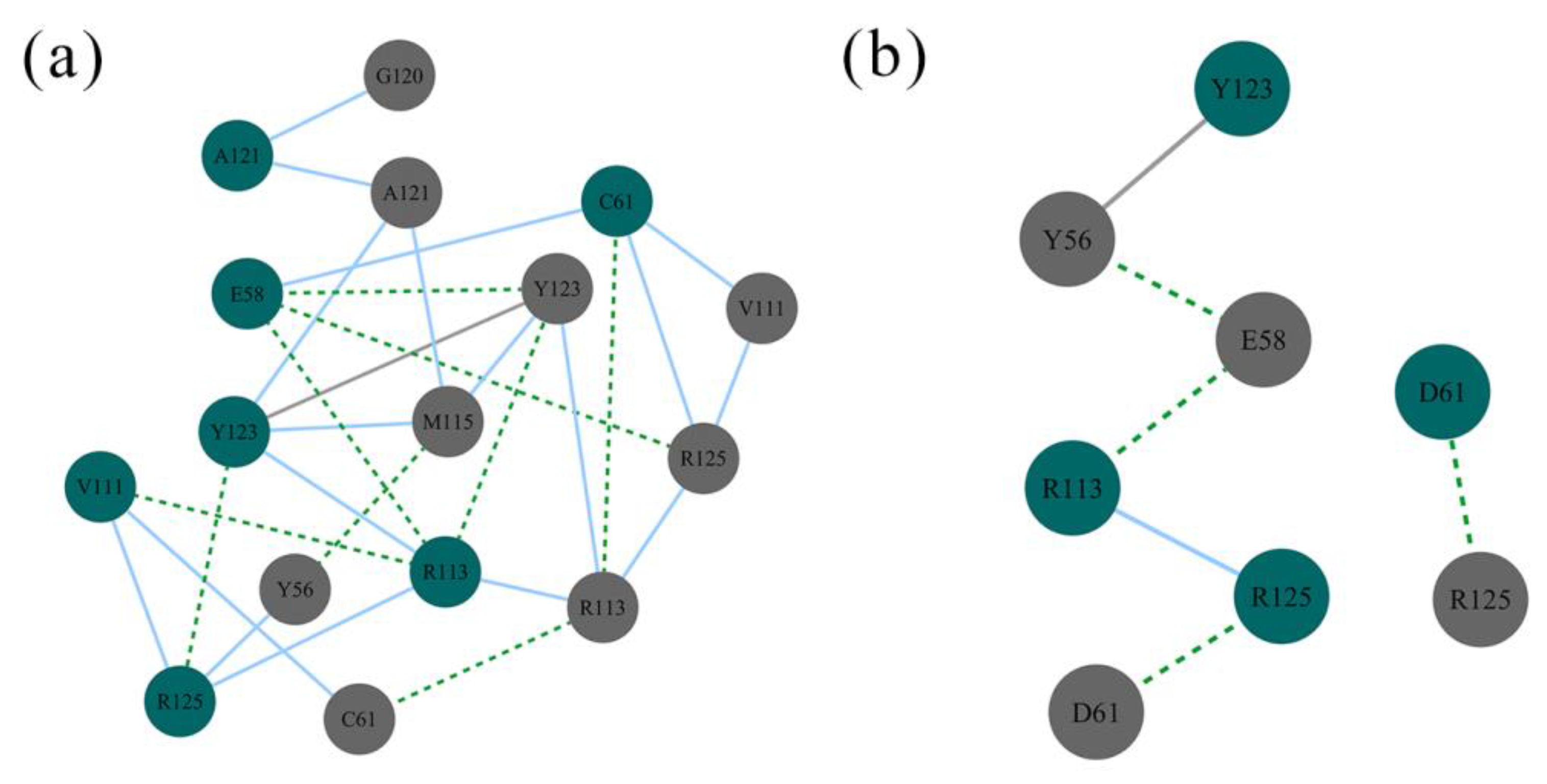
2.7. Nonbonded Interactions
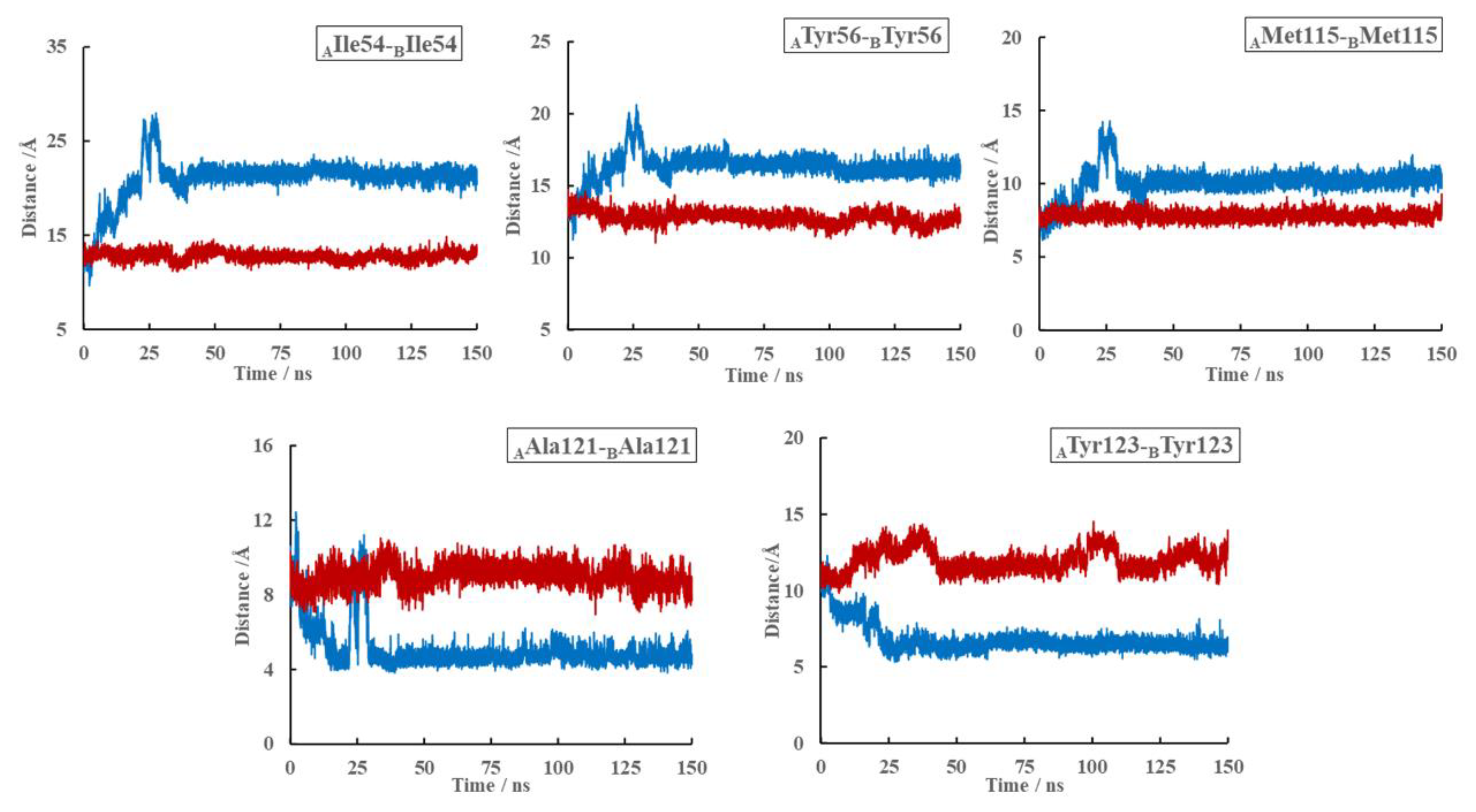
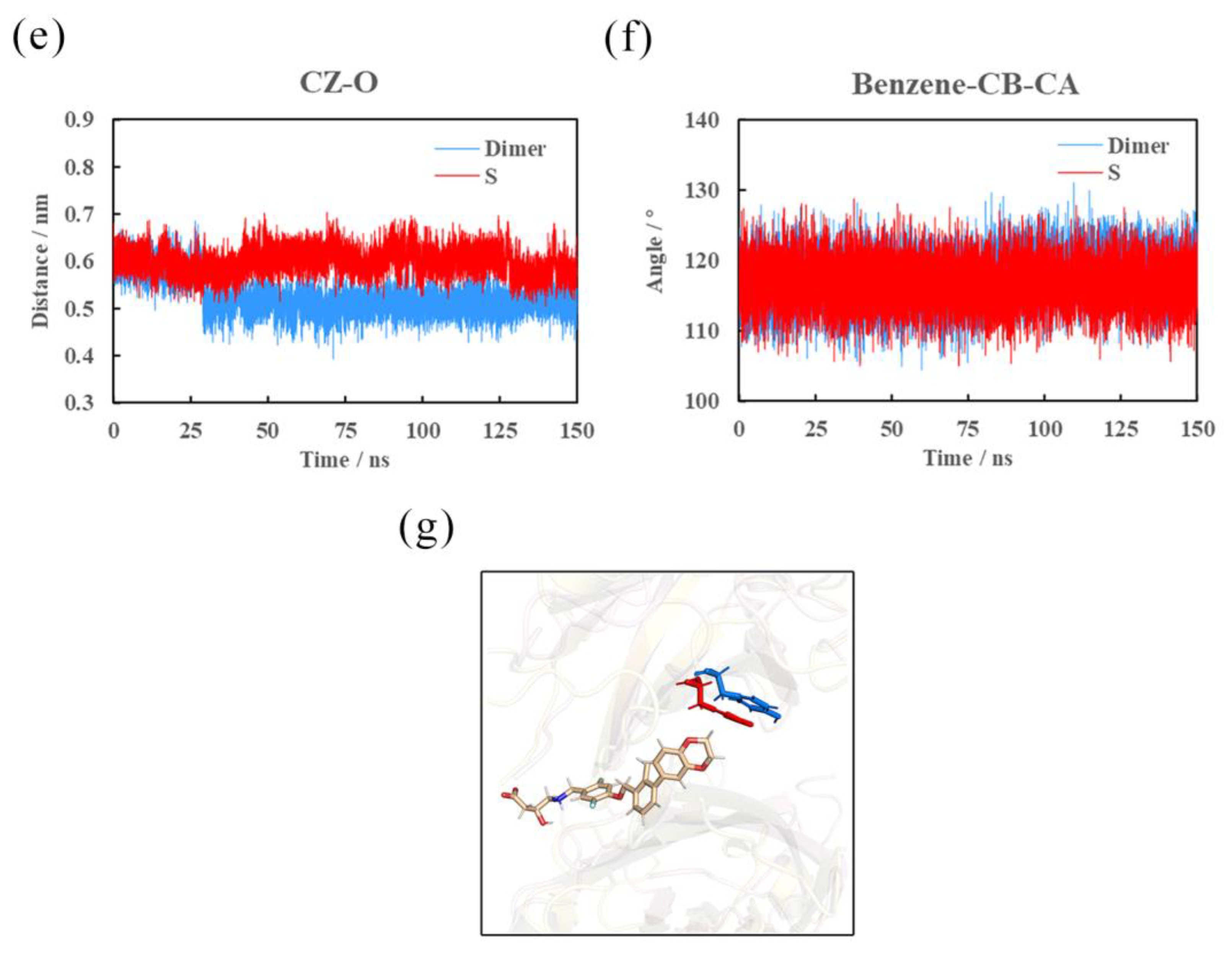
2.8. Effects of (S)-BMS-200 on the PD-L1 Dimer
3. Computational Details
3.1. Molecular Models
3.2. Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. Binding Free Energy Calculation
3.5. Simulation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (S)-BMS-200 | S-enantiomer of BMS-200 |
| (R)-BMS-200 | R-enantiomer of BMS-200 |
| (MOD)-BMS-200 | replacing the hydroxyl of (S)-BMS-200 with carbonyl |
| S system | PD-L1 dimer/(S)-BMS-200 |
| R system | PD-L1 dimer/(R)-BMS-200 |
| MOD system | PD-L1 dimer/(MOD)-BMS-200 |
| PD-L1 | programmed cell death ligand-1 |
| PD-1 | programmed cell death-1 |
| mAbs | monoclonal antibodies |
| MW | molecular weight |
| BMS | Bristol–Myers Squibb |
| PDB | Protein Data Bank |
| MD | molecular dynamics |
| MM-PBSA | molecular mechanics–Poisson Bolzmann surface area |
| APD-L1 | PD-L1 chain A |
| BPD-L1 | PD-L1 chain B |
| FEL | free energy landscape |
| PPI | protein–protein interaction |
| H bond | hydrogen bond |
| GAFF | general Amber force field |
| PME | particle mesh Ewald |
| Egas | gas-phase energy |
| Gsol | solvation-free energy |
| Evdw | van der Waals energy |
| Eele | electrostatic energy |
| EPB | polar solvation energy |
| ESA | nonpolar solvation energy |
| ΔS | entropy change |
| VMD | visual molecular dynamics |
| PCA | principal component analysis |
| RMSD | root mean square deviation |
| PLIP | Protein–Ligand Interaction Profiler |
| RMSF | root mean square fluctuation |
| PC | principal component |
References
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell co-stimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2016, 355, 1–14. [Google Scholar]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and function of the PD-L1 checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumorinfiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. PNAS 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Marzeca, M.; Zhanga, Q.; Goradiaa, A.; Raghunatha, P.N.; Liua, X.; Paesslera, M.; Wanga, H.Y.; Wysockab, M.; Cheng, M.; Ruggeric, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta. Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Philips, G.K.; Atkins, M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int. Immunol. 2015, 27, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug. Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Almahmoud, S.; Zhong, H.A. Molecular modeling studies on the binding mode of the PD-1/PD-L1 complex inhibitors. Int. J. Mol. Sci. 2019, 20, 4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmann, H. Cancer immunotherapy: Selected targets and small-molecule modulators. ChemMedChem 2016, 11, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorg. Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery, C.; Lynn, A. G_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Wang, T.; Kuai, Z.; Qian, M.; Tian, X.; Zhang, X.; Yu, Y.; Wang, S.; Zhang, H.; Li, H.; et al. Exploration of binding and inhibition mechanism of a small molecule inhibitor of influenza virus H1N1 hemagglutinin by molecular dynamics simulation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Qin, Y.; Wu, Y.; Zhao, W.; Zhai, W.; Qi, Y.; Wang, C.; Gao, Y. The design of high affinity human PD-1 mutants by using molecular dynamics simulations (MD). Cell. Commun. Signal. 2018, 16, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Quan, L.; Lyu, J.; He, Z.; Wang, X.; Meng, J.; Zhao, Z.; Zhu, L.; Liu, X.; Li, H. Discovery of peptide inhibitors targeting human programmed death 1 (PD-1) receptor. Oncotarget 2016, 7, 64967–64976. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Wen, W.; Liu, X.; Li, Y.; Zhang, J.Z.H. Computational analysis of hot spots and binding mechanism in the PD-1/PD-L1 interaction. RSC Adv. 2019, 9, 14944–14956. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Barakat, K. The too many faces of PD-L1: A comprehensive conformational analysis study. Biochemistry 2017, 56, 5428–5439. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yan, X.; Zhuo, W.; Gu, J.; Zuo, K.; Liu, W.; Liang, L.; Gan, Y.; He, G.; Wan, H.; et al. PD-L1 nanobody competitively inhibits the formation of the PD-1/PD-L1 complex: Comparative molecular dynamics simulations. Int. J. Mol. Sci. 2018, 19, 1984. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Zhou, S.; Liu, X.; Zhao, C.; Liu, H.; Yao, X. Understanding the structural and energetic basis of PD-1 and monoclonal antibodies bound to PD-L1: A molecular modeling perspective. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 576–588. [Google Scholar] [CrossRef]
- Shi, D.; An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Liu, H.; Yao, X. Computational insight into the small molecule intervening PD-L1 dimerization and the potential structure-activity relationship. Front. Chem. 2019, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, N.P.D., Jr.; Ager, L.A.; Bartlett, M.S.; Yardley, V.; Croft, S.L.; Khan, I.A.; McChesney, J.D.; Walker, L.A. Antiparasitic activities and toxicities of individual enantiomers of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-methyl-5-[3,4 dichlorophenoxy]quinol ine succinate. Antimicrob. Agents Chemother. 2008, 52, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.; Chun, J.; Jin, X.; Kim, J.; Yoon, J.; No, K.T. Investigation of protein-protein interactions and hot spot region between PD-1 and PD-L1 by fragment molecular orbital method. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Ma, S.; Liu, F.; Sun, Y.; Dong, X. Hematoxylin inhibits amyloid beta-protein fibrillation and alleviates amyloid-induced cytotoxicity. J. Phys. Chem. B 2016, 120, 11360–11368. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liang, L.; Gu, J.; Zhuo, W.; Yan, X.; Xie, T.; Wu, Z.; Liu, X.; Gou, X.; Liu, W.; et al. Inhibition of programmed cell death protein ligand-1 (PD-L1) by benzyl ether derivatives: Analyses of conformational change, molecular recognition and binding free energy. J. Biomol. Struct. Dyn. 2019, 37, 4801–4812. [Google Scholar] [CrossRef]
- Zhan, D.; Guan, S.; Jin, H.; Han, W.; Wang, S. Stereoselectivity of phosphotriesterase with paraoxon derivatives: A computational study. J. Biomol. Struct. Dyn. 2016, 34, 600–611. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Domling, A.; Dubin, G.; Holak, T.A. Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Forlemu, N.; Watkins, P.; Sloop, J. Molecular docking of selective binding affinity of sulfonamide derivatives as potential antimalarial agents targeting the glycolytic enzymes: GAPDH, aldolase and TPI. Open J. Biophys. 2017, 7, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 3, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Chang, H.W.; Chung, F.S.; Yang, C.N. Molecular modeling of p38alpha mitogen-activated protein kinase inhibitors through 3D-QSAR and molecular dynamics simulations. J. Chem. Inf. Model. 2013, 53, 1775–1786. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. Explicit ligand hydration shells improve the correlation between MM-PB/GBSA binding energies and experimental activities. J. Chem. Theory Comput. 2013, 9, 2706–2717. [Google Scholar] [CrossRef]
- Qin, B.; Jiang, X.; Lu, H.; Tian, X.; Barbault, F.; Huang, L.; Qian, K.; Chen, C.H.; Huang, R.; Jiang, S.; et al. Diarylaniline derivatives as a distinct class of HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2010, 53, 4906–4916. [Google Scholar] [CrossRef] [Green Version]
- Dragic, T.; Trkola, A.; Thompson, D.A.D.; Cormier, E.G.; Kajumo, F.A.; Maxwell, E.; Lin, S.W.; Ying, W.; Smith, S.O.; Sakmar, T.P. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. PNAS 2000, 97, 5639–5644. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ning, L.; Niu, Y.; Liu, H.; Yao, X. Molecular mechanism of the inhibition and remodeling of human islet amyloid polypeptide (hIAPP1-37) oligomer by resveratrol from molecular dynamics simulation. J. Phys. Chem. B 2015, 119, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 2005, 58, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sittel, F.; Jain, A.; Stock, G. Principal component analysis of molecular dynamics: On the use of cartesian vs. internal coordinates. J. Chem. Phys. 2014, 141, 1–10. [Google Scholar] [CrossRef]
- Riccardi, L.; Nguyen, P.H.; Stock, G. Free-energy landscape of RNA hairpins constructed via dihedral angle principal component analysis. J. Phys. Chem. B 2009, 113, 16660–16668. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Minervini, G.; Tosatto, S.C. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids. Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contribution | MOD | S | R | Dimer | APD-L1/ (S)-BMS-200 |
|---|---|---|---|---|---|
| ΔEvdw a | −70.70 ± 0.40 | −69.12 ± 0.59 | −67.93 ± 0.99 | −44.59 ± 9.85 | −35.75 ± 2.02 |
| ΔEele b | −12.88 ± 1.00 | −9.96 ± 0.78 | −12.15 ± 0.56 | −124.35 ± 23.36 | −6.33 ± 0.99 |
| ΔEPB c | 47.06 ± 1.38 | 42.55 ± 1.46 | 45.54 ± 1.81 | 211.28 ± 17.07 | 25.67 ± 2.70 |
| ΔESA d | −5.92 ± 0.04 | −5.88 ± 0.06 | −5.95 ± 0.04 | −6.23 ± 0.37 | −3.78 ± 0.18 |
| ΔEpolar,total e | 34.17 ± 2.38 | 32.59 ± 2.24 | 33.39 ± 2.37 | 86.94 ± 10.00 | 19.34 ± 0.92 |
| ΔEnonpolar,total f | −76.62 ± 0.44 | −75.00 ± 0.65 | −73.88 ± 1.03 | −50.82 ± 10.06 | −39.53 ± 3.68 |
| ΔG g | −42.45 ± 0.35 | −42.42 ± 0.21 | −40.48 ± 0.21 | 36.11 ± 0.89 | −20.17 ± 2.20 |
| Inhibitor | N-Terminal | Loop | Total Sheet | Total |
|---|---|---|---|---|
| (S)-BMS-200 | 33 | 18 | 457 | 507 |
| (R)-BMS-200 | 22 | 24 | 446 | 492 |
| Donor | Donor H | Acceptor | Occupancy (%) |
|---|---|---|---|
| APhe19@N | H | (S)-BMS-200@O5 | 28.57 |
| APhe19@N | H | (S)-BMS-200@O4 | 22.92 |
| APhe19@N | H | (S)-BMS-200@O3 | 0.33 |
| BHis69@NE2 | HE2 | (S)-BMS-200@O5 | 1.33 |
| BHis69@NE2 | HE2 | (S)-BMS-200@O4 | 1.33 |
| BHis69@NE2 | HE2 | (S)-BMS-200@O3 | 1.33 |
| Donor | Donor H | Acceptor | Occupancy (%) |
|---|---|---|---|
| (R)-BMS-200@O3 | H3 | AAsp122@OD2 | 42.52 |
| ALys124@NZ | HZ2 | (R)-BMS-200@O4 | 4.98 |
| ALys124@NZ | HZ3 | (R)-BMS-200@O5 | 2.99 |
| BLys75@NZ | HZ2 | (R)-BMS-200@O4 | 1.66 |
| BGln66@NE2 | 2HE2 | (S)-BMS-200@O5 | 0.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. https://doi.org/10.3390/ijms22094766
Guo Y, Jin Y, Wang B, Liu B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. International Journal of Molecular Sciences. 2021; 22(9):4766. https://doi.org/10.3390/ijms22094766
Chicago/Turabian StyleGuo, Yan, Yulong Jin, Bingfeng Wang, and Boping Liu. 2021. "Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization" International Journal of Molecular Sciences 22, no. 9: 4766. https://doi.org/10.3390/ijms22094766
APA StyleGuo, Y., Jin, Y., Wang, B., & Liu, B. (2021). Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. International Journal of Molecular Sciences, 22(9), 4766. https://doi.org/10.3390/ijms22094766