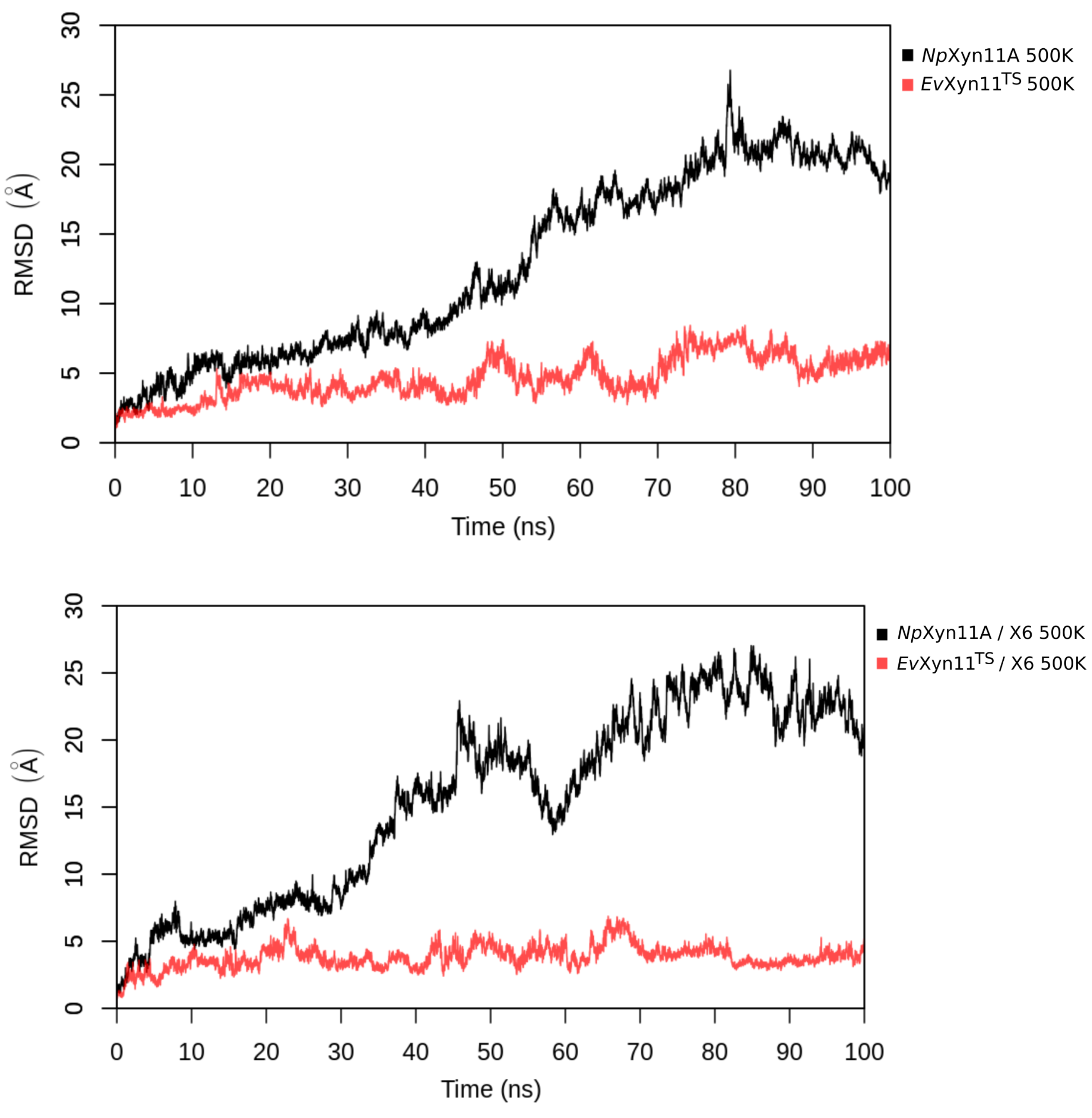
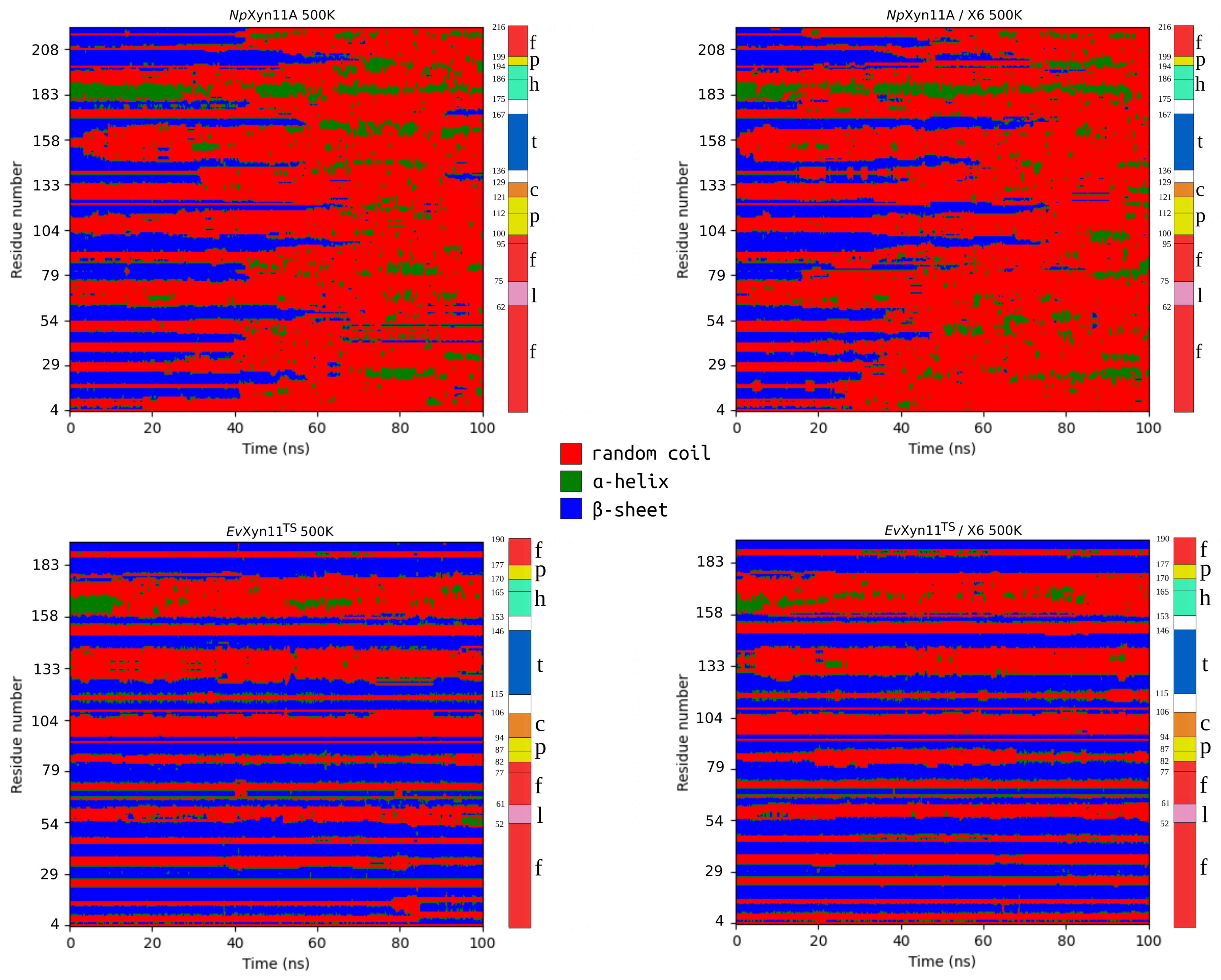
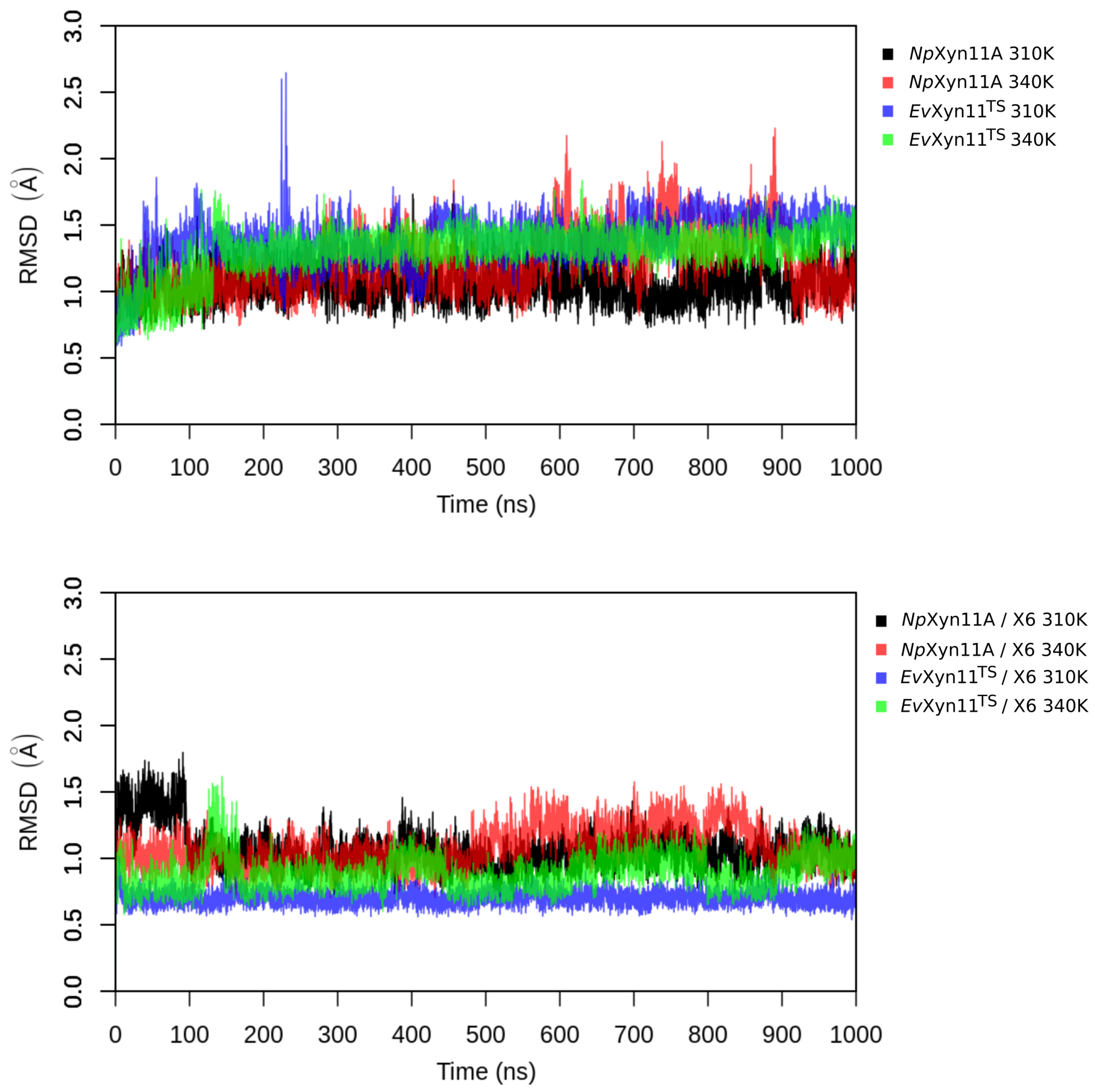
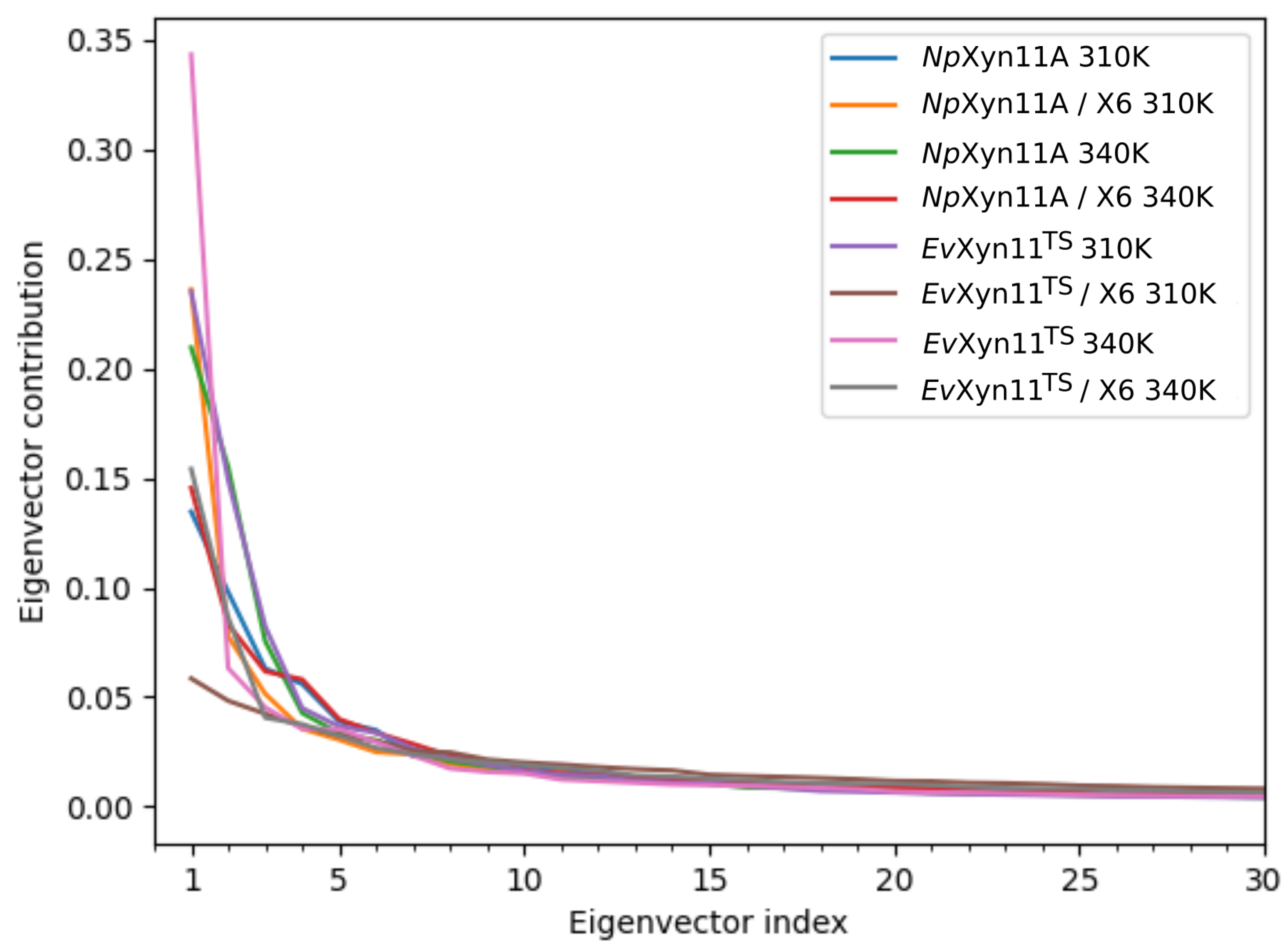
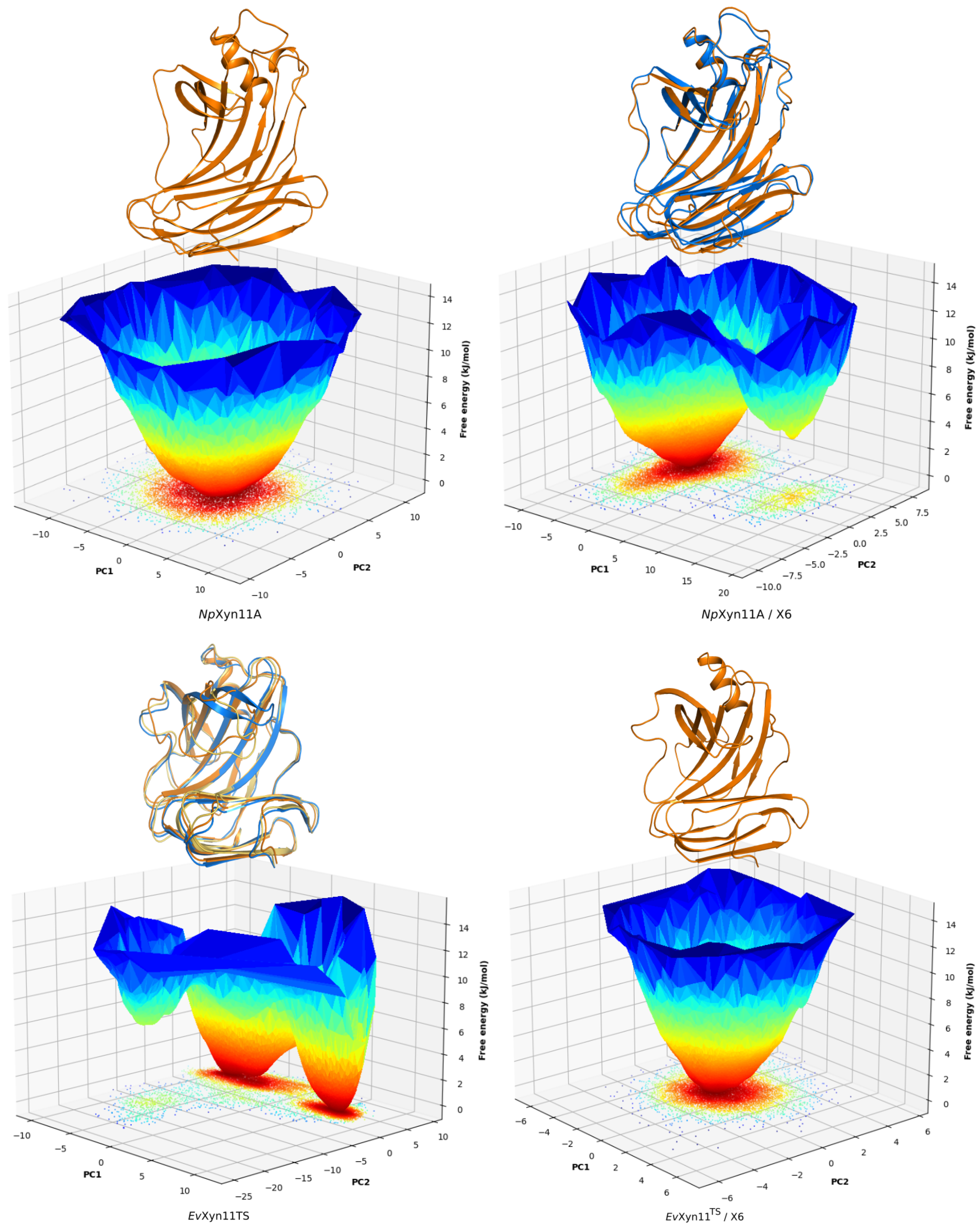
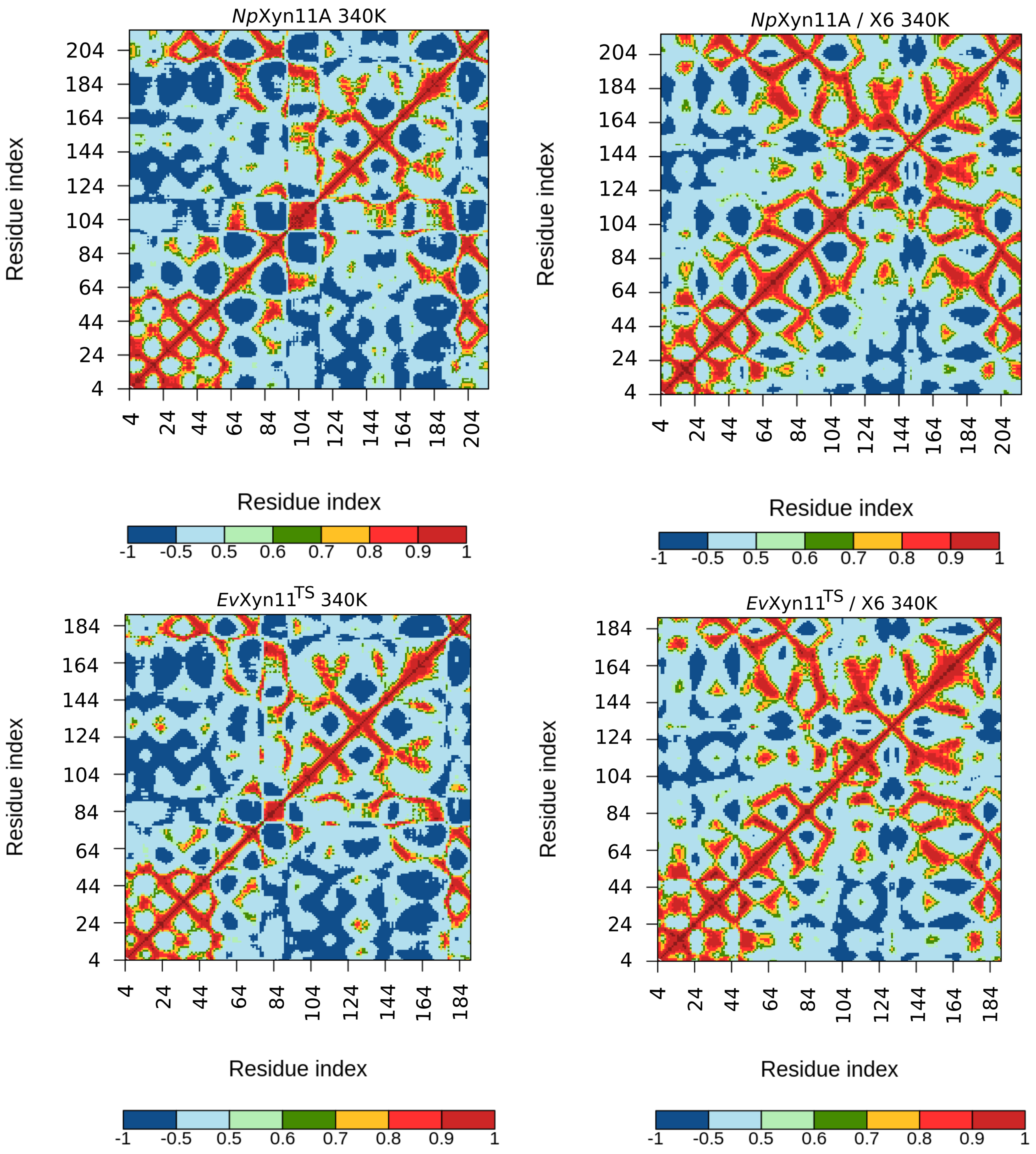
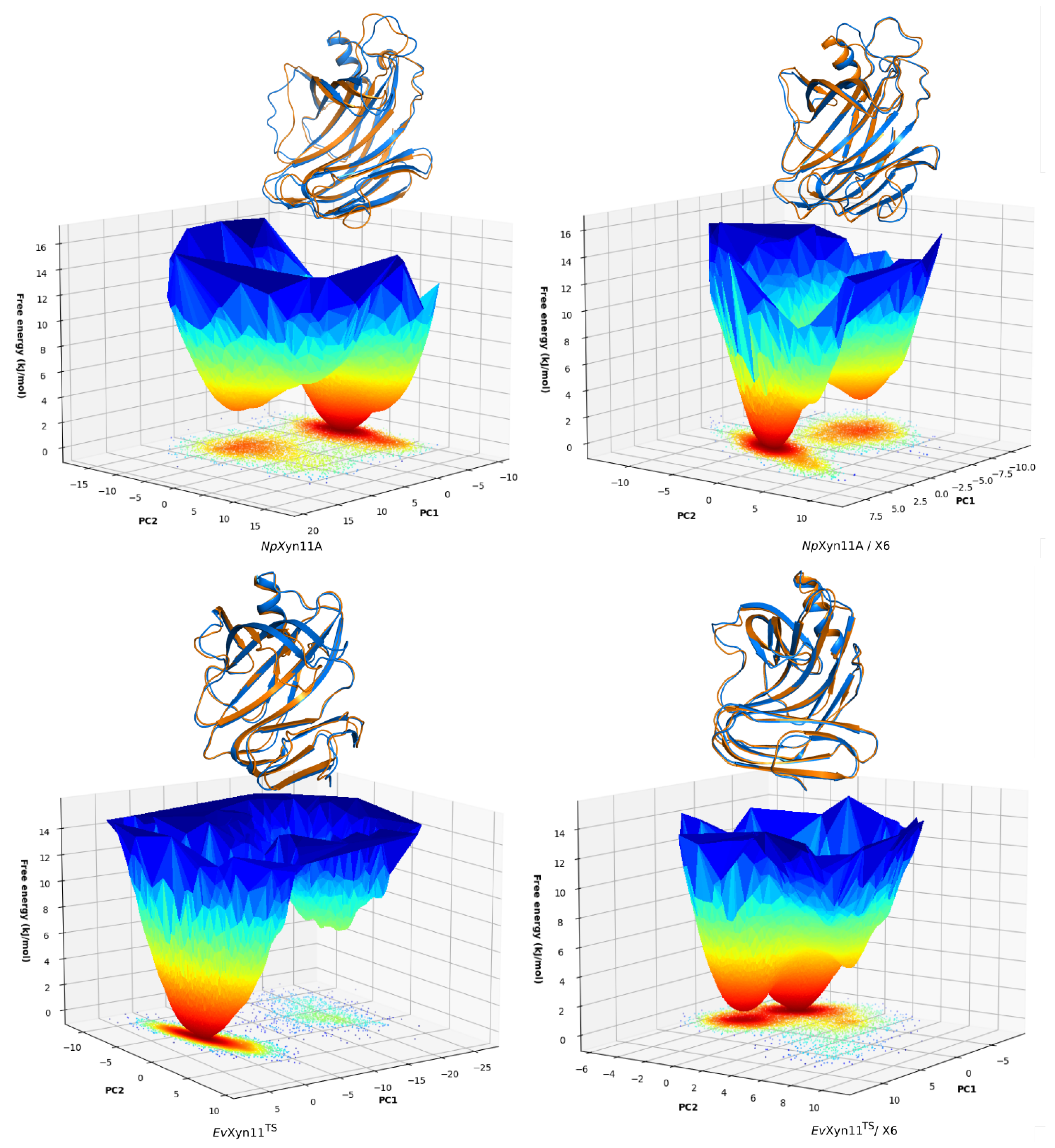
PCA analysis was based on the first two principal components that explain, on average, 30% of the total protein motions. The contribution of the first principal component varies between 5% and 35%. The minimal value of 5% corresponds to the simulation performed on EvXyn11TS in complex with xylohexaose at 310 K. This system showed very low B-factor values in the simulations resulting from the noise that spreads equally along all axis.
3.5.1. Hydrogen Bonds, Salt Bridges, and SASA
The static and dynamic intramolecular and enzyme–solvent hydrogen bond (HB) counts in
NpXyn11A and
EvXyn11
TS in their free-enzyme and enzyme–substrate complex forms were computed from MD simulations at 310 K and 340 K (
Table 2).
EvXyn11
TS has more static hydrogen bonds, which contribute to the formation of larger stabilizing interaction networks.
NpXyn11A instead possesses a higher number of dynamic HBs, which reflect the dynamic formation of competitive HB interactions. Furthermore, the total number of enzyme–solvent hydrogen bonds observed during 1 μs MD simulation is also higher for
NpXyn11A. The transient existence of a greater number of dynamic HBs in
NpXyn11A and with the solvent is consistent with its greater flexibility.
Salt bridges have been identified as one of the main factors contributing to thermostability within the GH-11 family [
16,
46]. We monitored the different salt bridges that are formed within the protein structures over 1 μs of simulation time (
Table 3). The three-dimensional structures of the identified salt bridges in the free-enzymes are shown in
Figure 10. In the enzyme–xylohexaose complexes, the same salt bridges as those observed in the unbound enzymes were detected, except the ones involving a catalytic residue, Glu201 (the catalytic acid–base residue in
NpXyn11A) or Glu89 (the catalytic nucleophile residue in
EvXyn11
TS), given that these residues interact with the substrate in the complex.
A total of 8 salt bridges stabilize the NpXyn11A enzyme, against the 4 in the highly thermostable EvXyn11TS. However, salt bridges are present in 33 to 98% of the total simulation time for NpXyn11A, against 94 to 99% of the total simulation time for EvXyn11TS.
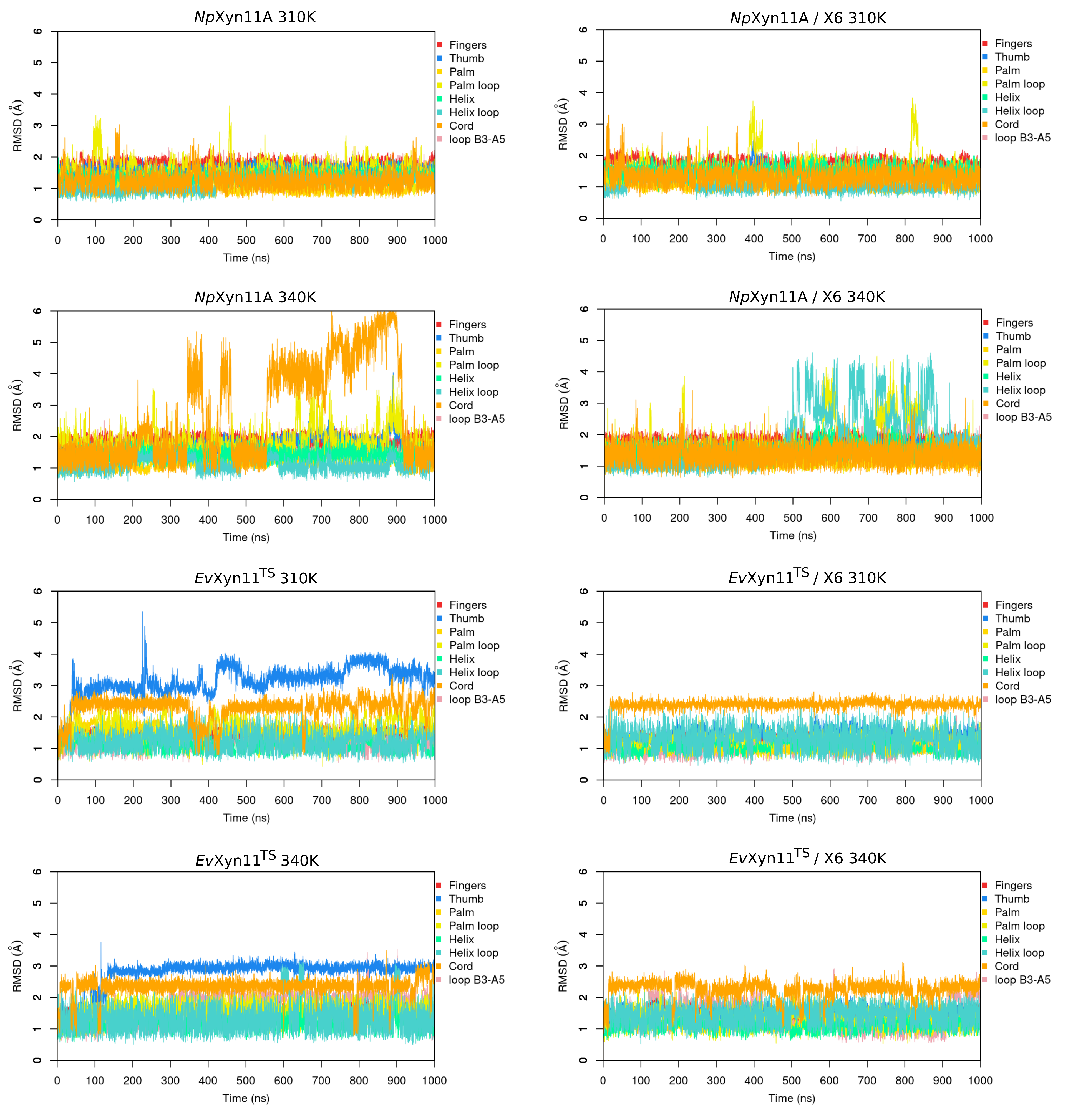
In NpXyn11A, a relatively stable salt bridge (80% occurrence frequency) is formed by Asp145–Lys159 in the important thumb region. This may explain the moderate flexibility of this region in NpXyn11A at both studied temperatures and suggests that in both bound and unbound forms, NpXyn11A possesses a stable thumb conformation, which is probably already competent for catalysis. On the contrary, the absence of an intrathumb salt bridge in EvXyn11TS can explain the high flexibility of this region observed in this enzyme.
In
NpXyn11A, another salt bridge is formed by the Asp126–Lys140 pair. Located between the cord and the thumb region, this salt bridge becomes less frequent at 340 K, which is consistent with the increased backbone flexibility of the cord region in the free-enzyme form of
NpXyn11A at 340 K (
Table 3). Analysis of salt bridges also reveals that the stable Glu118–Arg169 (in
NpXyn11A) and Asp94–Arg149 (in
EvXyn11
TS) interactions occupy the same location when superimposing the structures, meaning that these salt bridges are conserved in both xylanases.
In
NpXyn11A, an intrahelix salt bridge is also found between the residues Glu182 and Lys185 with an occurrence frequency of 73%. Other salt bridges were detected with a lower occurrence frequency, especially the Glu22–Lys13 (45% occurrence frequency) and the Lys42–Asp210 (56% occurrence frequency) salt bridges located between the fingers. These salt bridges are less stable than the Arg38–Asp190 salt bridge (99% occurrence frequency) found in
EvXyn11
TS fingers. The presence of both this salt bridge and a disulfide bridge must strongly contribute to the high stability of the
N-terminal and fingers regions of
EvXyn11
TS. In
EvXyn11
TS, a very stable (98% frequency) salt bridge, formed by the Asp161–Arg60 pair between the
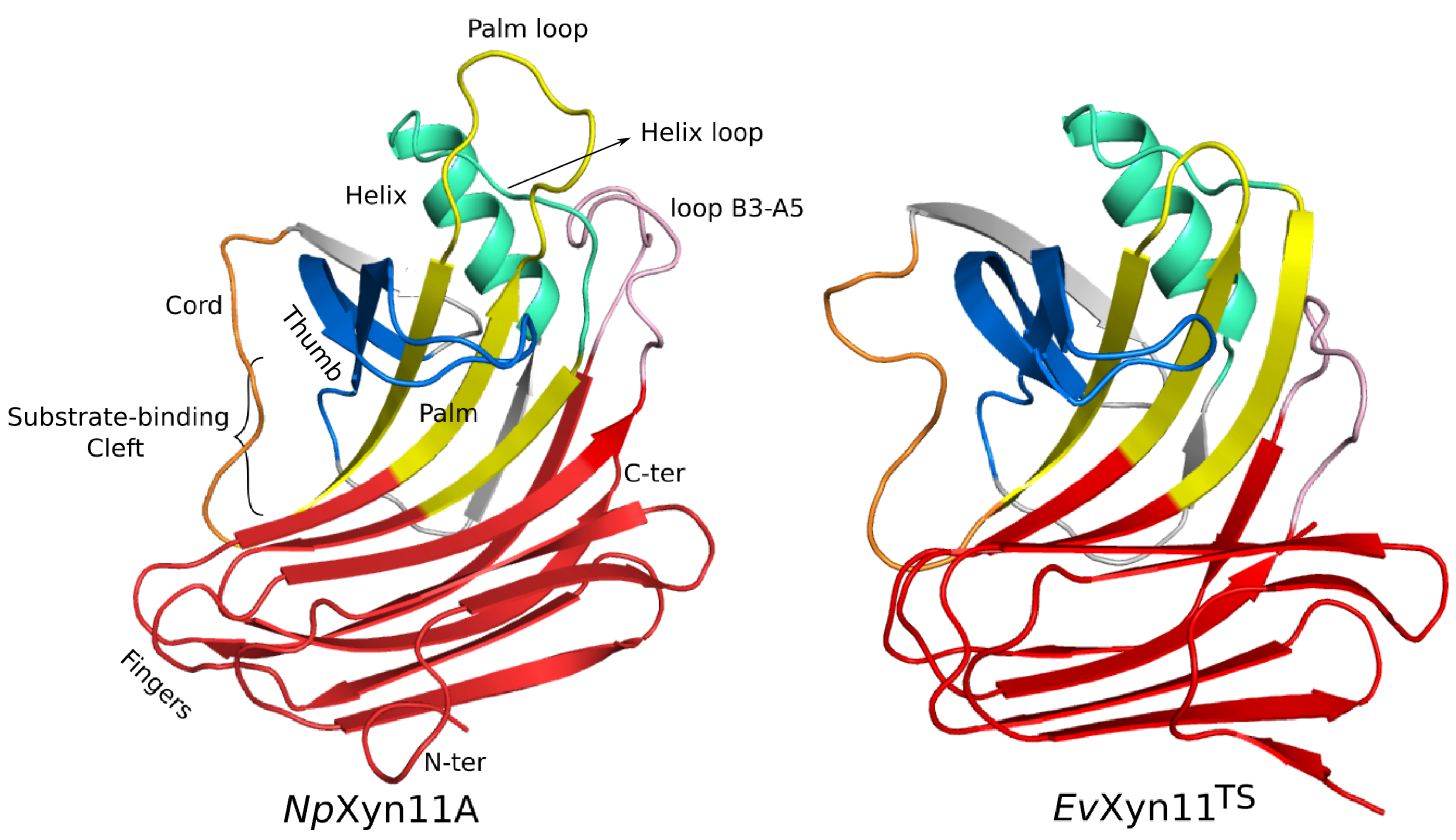
-helix and the B3-A5 loop (in pink in
Figure 1) must also contribute to the global stability of the enzyme by stabilizing the interaction between two distinct regions of the 3D structure involving a surface loop.
Overall, the formation of very stable salt bridges in EvXyn11TS probably plays an essential role for structural maintenance and, consequently, for the enhanced thermostability of the protein.
Finally,
Table 4 shows the average Surface Accessible Solvent Areas for each enzyme over the course of their MD trajectories.
NpXyn11A shows higher SASA than
EvXyn11
TS, which is consistent with the higher number of enzyme–solvent hydrogen bonds detected in
NpXyn11A Ȧll of these analyses indicate that the mesostable
NpXyn11A enzyme is less tightly packed than the hyper-thermostable
EvXyn11
TS enzyme, and possesses a lower number of static intrahydrogen bonds and less-stable salt bridges.
3.5.2. Analysis of Enzyme/Substrate Interactions
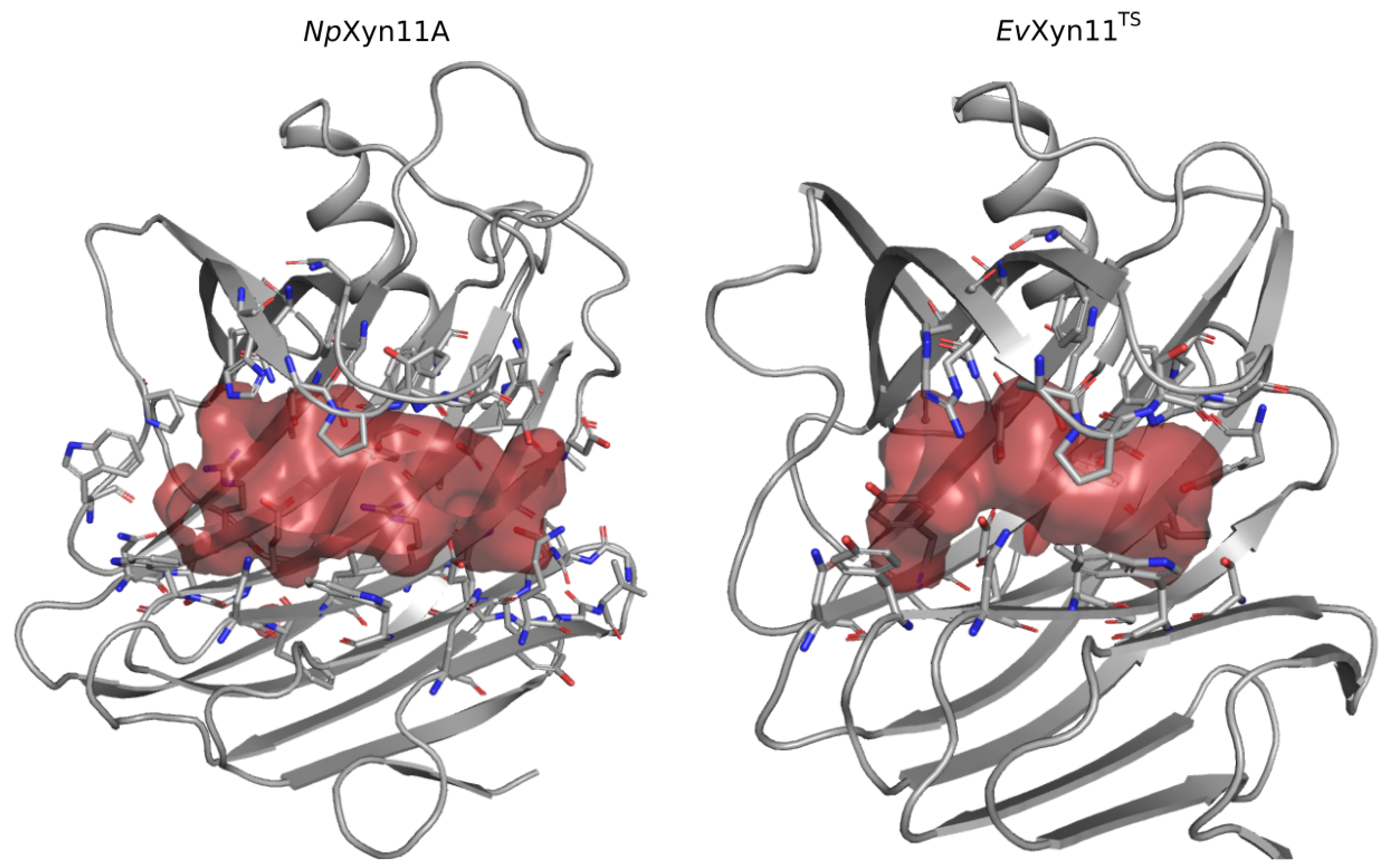
One of the main features of the globular structure of these enzymes is the presence of a long cleft located in the center of the enzyme, which contains the active site (also shown in
Figure 1). The active site of each enzyme was analyzed in terms of residue composition, negative volume, and area of the open cleft.
Figure 11 shows the residues that compose each enzyme’s active site as well as its corresponding negative volume (summarized in
Table 5 with the corresponding area).
In the center of the active site of each enzyme are the two conserved catalytic residues, the catalytic nucleophile (Glu113 in
NpXyn11A and Glu 89 in
EvXyn11
TS) and the catalytic acid–base (Glu201 in
NpXyn11A and Glu181 in
EvXyn11
TS). Moreover, several other amino acids in the binding site of both enzymes are highly conserved in GH-11 xylanases [
3,
47], including Ile151 in
NpXyn11A (Ile132 in
EvXyn11
TS) at −3 subsite; Tyr98 (Tyr80
EvXyn11
TS) and Trp100 (Trp82
EvXyn11
TS) at −2 subsite; Pro149 (Pro130
EvXyn11
TS) and Phe158 (Phe138
EvXyn11
TS) at −1 subsite; Tyr115 (Tyr91
EvXyn11
TS) and Gln160 (Gln140
EvXyn11
TS) at +1/−1 subsites; and Pro125 (Pro101
EvXyn11
TS) at +1 subsite. Two additional highly conserved amino acid residues are found in the binding site of
EvXyn11
TS: Ser131 at the −3 subsite and Arg126 at the −1 subsite. In
NpXyn11A, the corresponding positions are occupied by Thr150 and His146.
The active site cleft (AS) is almost six times larger in
NpXyn11A than in
EvXyn11
TS. It encompasses 41 residues with a volume of 461.24 Å
3 and an area of 521.62 Å
2, while the active site cleft of
EvXyn11
TS possesses only 23 residues, a volume of 77.12 Å
3, and an area of 173.27 Å
2. The larger and extended binding cleft of
NpXyn11A was shown to play an important role in the unusually high activity displayed by this enzyme [
17]. Indeed, these active site features may facilitate the access or release of the substrate or product while better accommodating xylose units in each of its subsites. Furthermore, the wider cleft of
NpXyn11A might also facilitate the recognition and interaction with decorated xylans and, consequently, their hydrolysis.
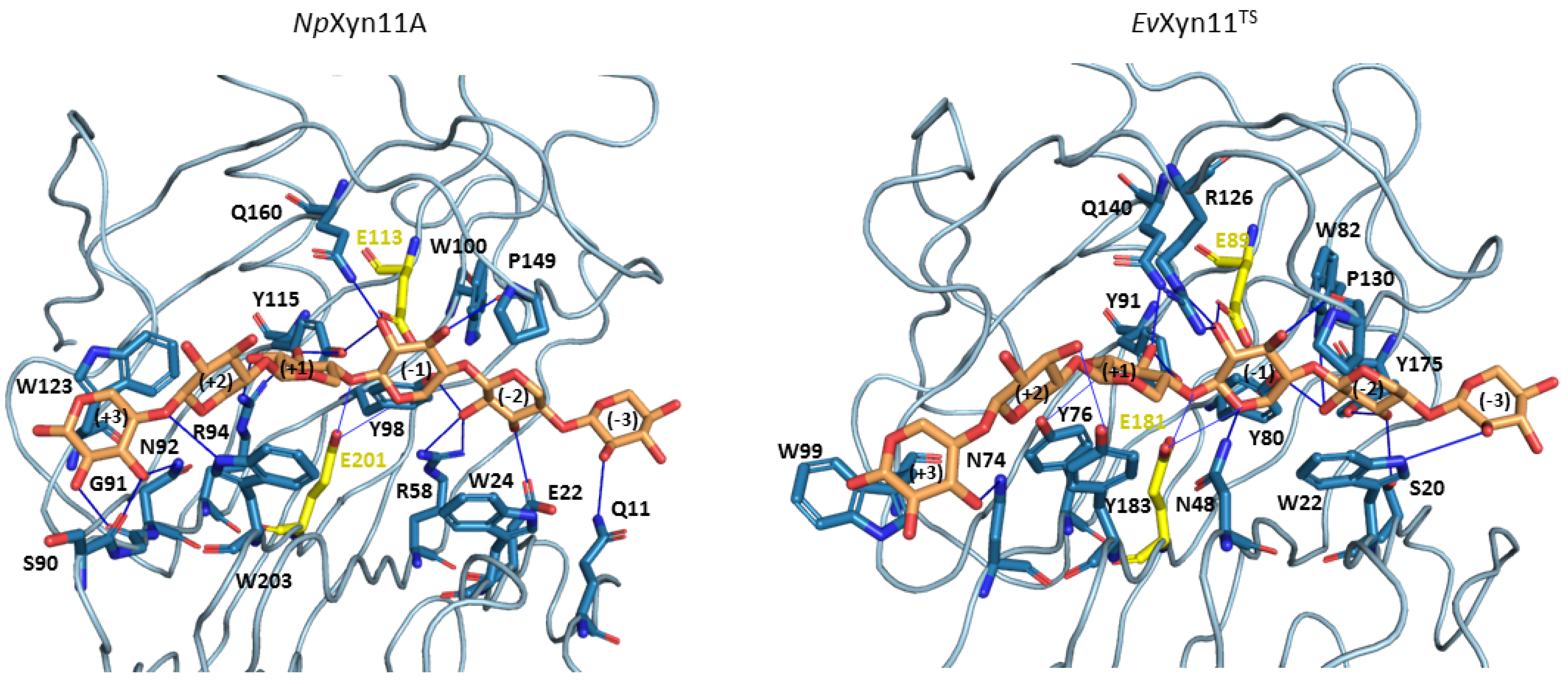
The xylohexaose (X6) adopts a similar conformation in the active site of both enzymes and is maintained in the binding cleft all along the MD simulations. As hydrogen bonds and stacking interactions provide the main ligand binding strengths, these noncovalent interactions have been evaluated using the PLIP web-server. To consider the most catalytically favorable conformation of xylohexaose in the binding site, we firstly chose to analyze the interactions in the 3D structure of
NpXyn11A/X6 and
EvXyn11
TS/X6 generated after the equilibration phase. Different interactions are presented in
Figure 12, where we display the initial equilibrated configuration of the respective enzymes in complex with xylohexaose. There is a total of 20 different hydrogen bonding interactions with xylohexaose in
NpXyn11A, versus 16 in
EvXyn11
TS. The list of residues involved in hydrogen-bonding interactions with X6 in each enzyme is given in
Table 6. These residues, distributed from −3 to +3 subsites of the binding cleft, include several highly conserved amino acid residues, previously mentioned (Tyr88, Trp100, Pro149, and Tyr115 in
NpXyn11A; Tyr80, Trp82, Pro130, and Tyr91 in
EvXyn11
TS). In addition to hydrogen bonds, the xylohexaose is also stabilized in the binding site by stacking interactions involving the aromatic amino acid residues: Trp24, Trp203, and Trp123 in
NpXyn11A; Trp22, Tyr183, and Trp99 in
EvXyn11
TS at the subsites −2, +2, and +3, respectively.
The density of the enzyme–xylohexaose hydrogen bond network is relatively similar in the central subsites (−2 to +2) of both enzymes. However, subsite +3 shows a sparser network in EvXyn11TS than in NpXyn11A. In NpXyn11A, the loop connecting the -strands at subsite +3 is 4 residues longer than in EvXyn11TS and allows more interactions with the xylose unit. The backbone carbonyl groups of Ser90 and Gly91 form hydrogen bonds with the hydroxyl groups of the xylose at subsite +3, in addition to the polar interaction involving the side chain of Asn92. In contrast, in EvXyn11TS, the loop is shorter, and only Asn74 makes a polar interaction with the xylose unit at subsite +3. While the side-chain of Gln10 (the first residue of a -strand in the N-terminal domain of EvXyn11TS) points away from the xylohexaose binding site, the side-chain of the corresponding Gln11 in NpXyn11A points towards the binding site and is perfectly oriented to make a polar contact with the xylose unit at subsite −3. The difference in the distal glycone (−3) and aglycone (+3) subsites of the extended cleft of NpXyn11A might explain the high catalytic activity displayed by this enzyme compared to EvXyn11TS.
To go further in the investigation of enzyme–substrate interactions, the intermolecular hydrogen bonds formed between the enzymes and the xylohexaose substrate were also monitored during the first 100 ns of the respective MD trajectories at 310 K.
Table 7 shows the frequency of occurrence of the intermolecular hydrogen bonds established between
NpXyn11A and xylohexaose, and between
EvXyn11
TS and xylohexaose, respectively, when this frequency exceeds 10%.
Over the course of the MD trajectories, we observe the formation of only one additional type of intermolecular hydrogen bond in each complex compared to the ones initially observed in the equilibrated conformations. Indeed, a hydrogen bond is formed between the Asn54 and the xylose unit at subsite +1 of the
NpXyn11A cleft (with an occurrence frequency of 65%) and between the Asp105 located in the cord region and the xylose subunit in the subsite +2 of the
EvXyn11
TS cleft (with a frequency of 28%) (
Table 7). In the initial
EvXyn11
TS structure, the Asp105 is located at a distance of 7 Å from the ligand. This observation confirms that a conformational rearrangement of this flexible region is required to allow this amino acid to interact with the substrate in the
EvXyn11
TS/X6 complex. It is probable that the binding of the ligand triggers this conformational change.
Some interactions, observed in the initial configurations, are present in less than 10% of the 100 ns MD of both NpXyn11A and EvXyn11TS. In NpXyn11A, this is especially the case for the side-chains of Arg58 and Trp100, which initially interact with the xylose unit at subsite −2; Gln160, which initially interacts with the xylose at subsite −1; Tyr115, which initially interacts with the xylose at subsites −1 and +1; and Arg94, which initially interacts with the xylose at subsite +1. In EvXyn11TS, this is the case for the side-chain of Trp22, which initially interacts with the xylose at subsite −3; Ser20 and Trp82, which initially interact with the xylose at subsite −2; Tyr91, which initially interacts with the xylose at subsite +1; and finally, Tyr183, which initially interacts with the xylose at subsite +2.
Compared with
NpXyn11A, the interaction of the substrate in the −3 subsite (glycone) is less stable in
EvXyn11
TS. As hydrogen interactions involving the residues of the distal glycone subsite (−3) are crucial for the binding of X6 substrate, the loss of interaction identified in
EvXyn11
TS might contribute to its lower catalytic activity. At subsite −2, the conserved tyrosine in GH-11 xylanases; Tyr98 and Tyr80 in
NpXyn11A and
EvXyn11
TS, respectively; in addition to Glu22 in
NpXyn11A and Tyr175 in
EvXyn11
TS, make polar interactions with the substrate along MD simulations with an occurrence frequency of 21%, 67%, 43% and 80% respectively. The lower occurrence frequency of these interactions in
NpXyn11A may be explained by the bigger
NpXyn11A active site cleft, especially in the glycone region, thus enabling the substrate to interact with more residues over the course of the simulation. Furthermore, the presence of a tryptophan residue at position 24 in
NpXyn11A and at position 22 in
EvXyn11
TS likely promotes a stacking interaction of the indole ring with the xylose moiety at subsite −2. At subsite −1, the highly conserved proline in GH-11 xylanases, found at position 149 in
NpXyn11A 130 in
EvXyn11
TS, and located in the thumb loop, is involved in forming a conserved pattern of HB interactions in more than 76% of both enzymes MD trajectories (
Table 7). Arg126 in
EvXyn11
TS, which is also located in the thumb region close to this proline in
EvXyn11
TS, makes a polar contact with the xylose unit at subsite −1 (
Figure 12). Histidine (His146) residue is found at this position in
NpXyn11A. It has been suggested that this residue could form polar contacts with surrounding residues [
17]. As observed in the equilibrated conformations, the substrate is better stabilized with more polar contacts at subsite +3 of
NpXyn11A than in
EvXyn11
TS. The substrate binding to the distal glycone and aglycone subsites is thus weaker in
EvXyn11
TS. By contrast, the topology of the
NpXyn11A cleft allows it to better accommodate and make more polar interactions with the xylose units at the −3 and +3 subsites, which may contribute to the highly enhanced activity of
NpXyn11A in comparison to
EvXyn11
TS.
The analyses of the unusually active NpXyn11A and hyper-thermostable EvXyn11TS in complex with xylohexaose revealed details on the molecular determinants playing a key role on xylohexaose binding interactions, which could be transposed to other xylanases in the GH-11 family.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}