
Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
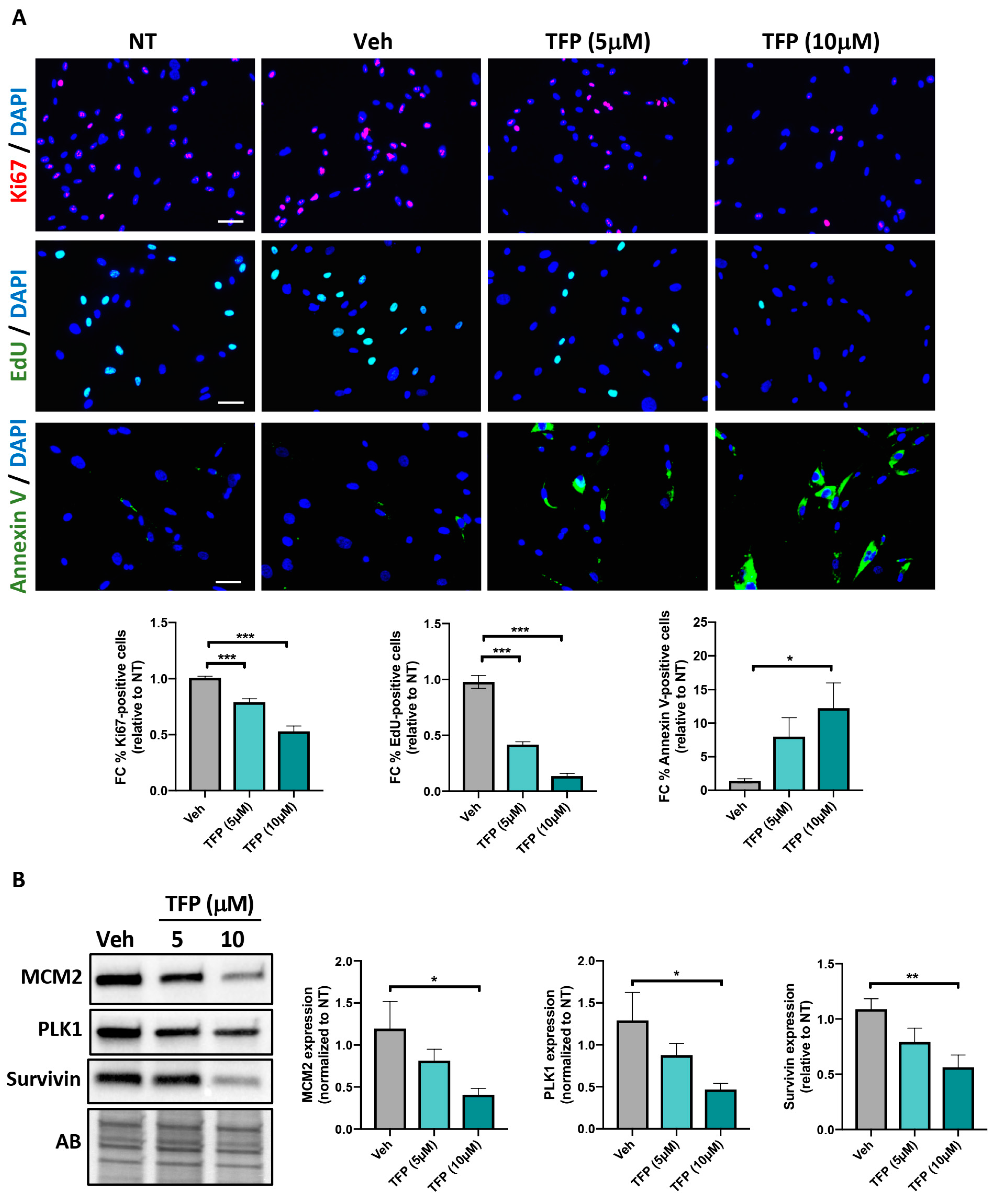
2.1. TFP Significantly Reduced PAH-PASMC Proliferation and Survival
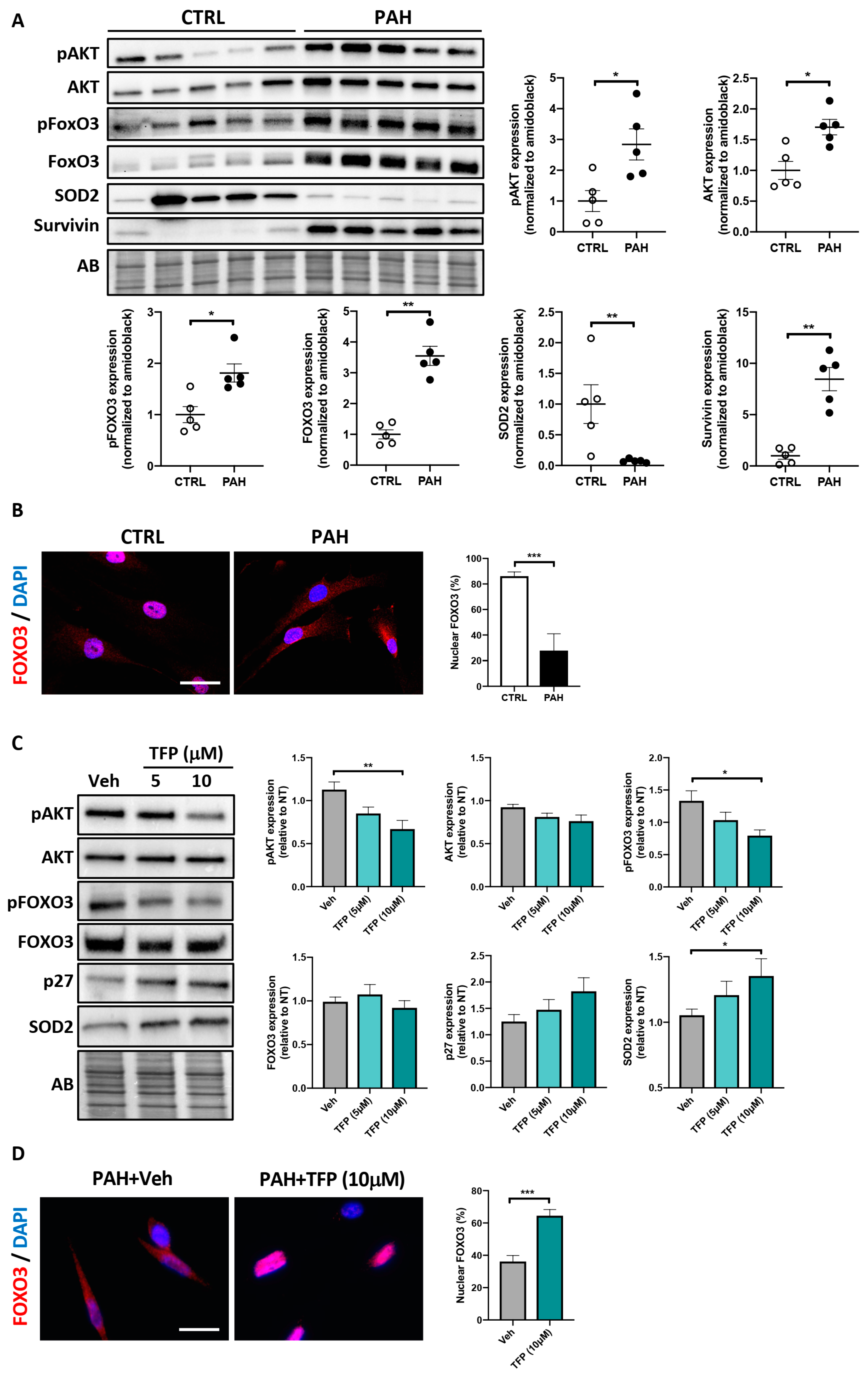
2.2. Effects of TFP on AKT/FOXO3 Signaling in PAH-PASMCs
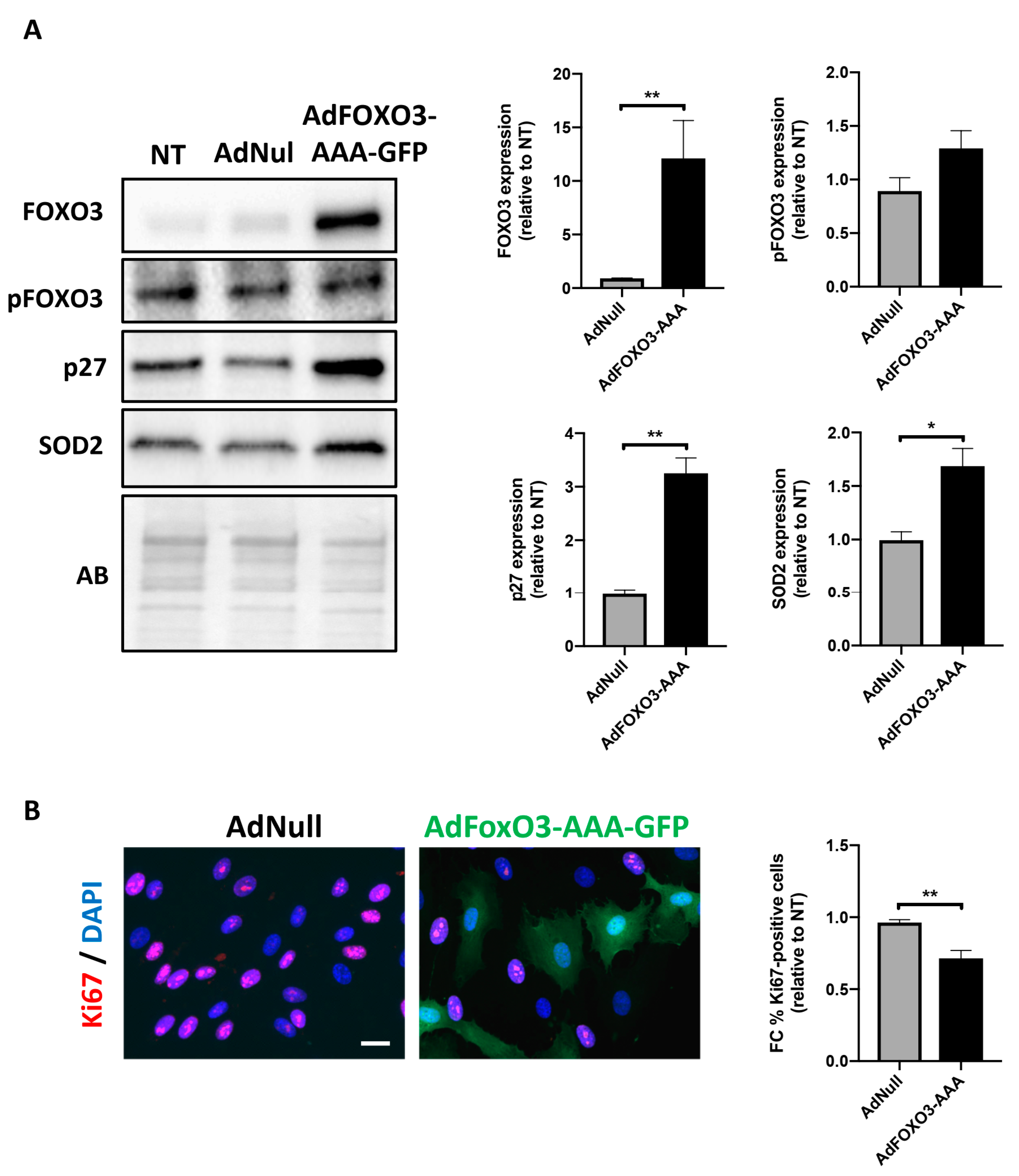
2.3. Forced Nuclear Localization of FOXO3 Reduces PAH-PASMC Proliferation
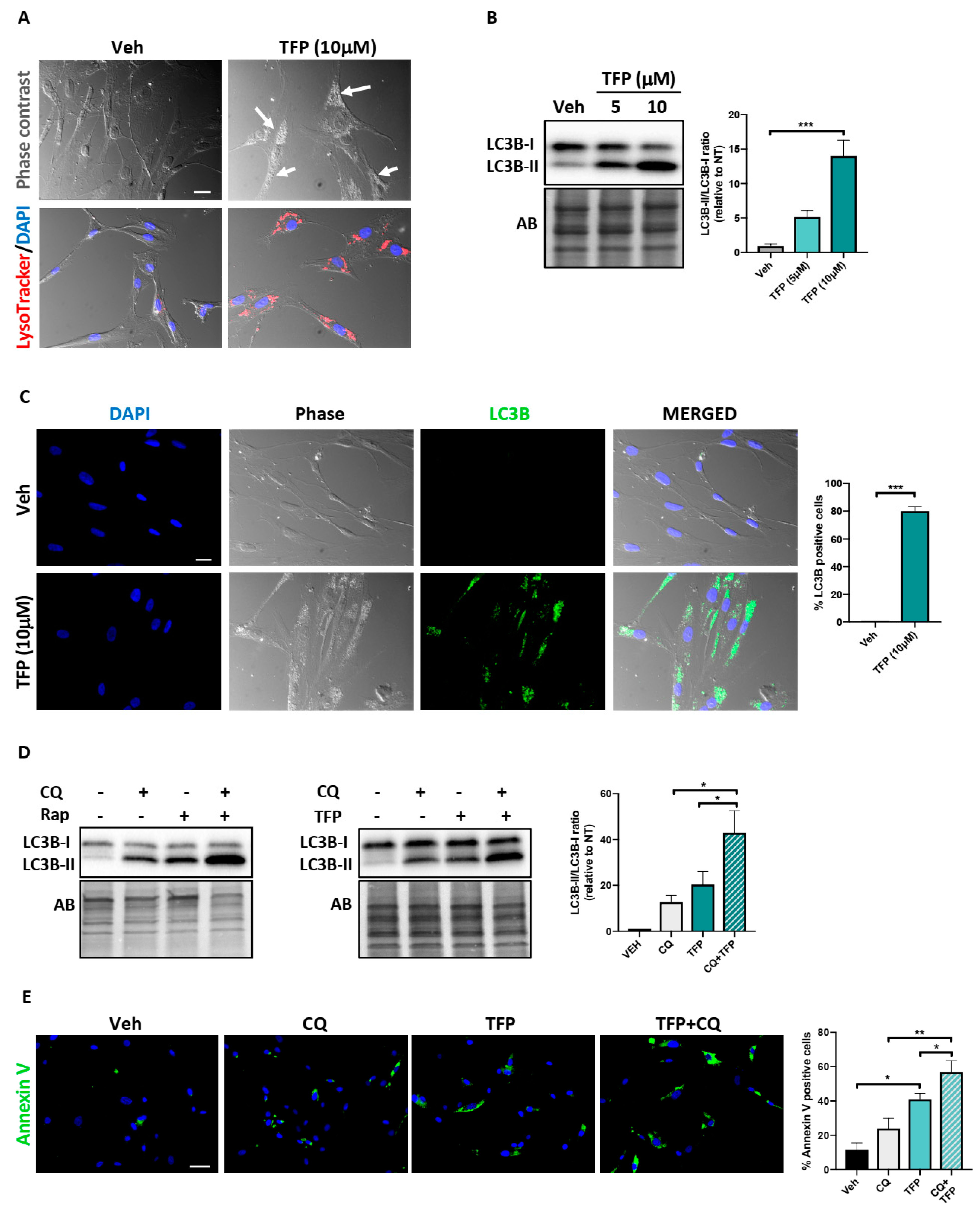
2.4. Trifluoperazine Induces Autophagy
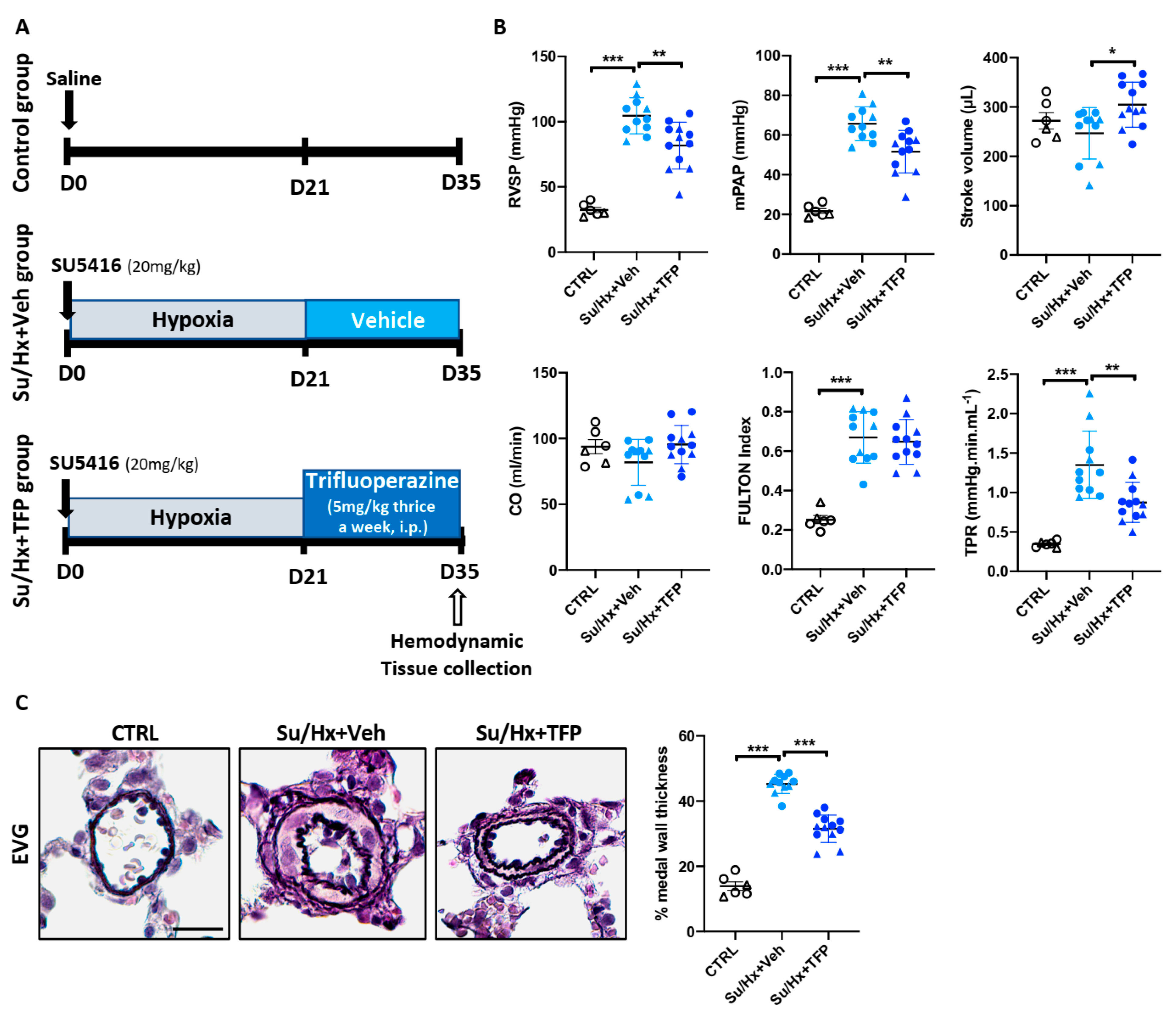
2.5. Trifluoperazine Significantly Improves Established PAH in the Sugen/Hypoxia (Su/Hx) Rat Model
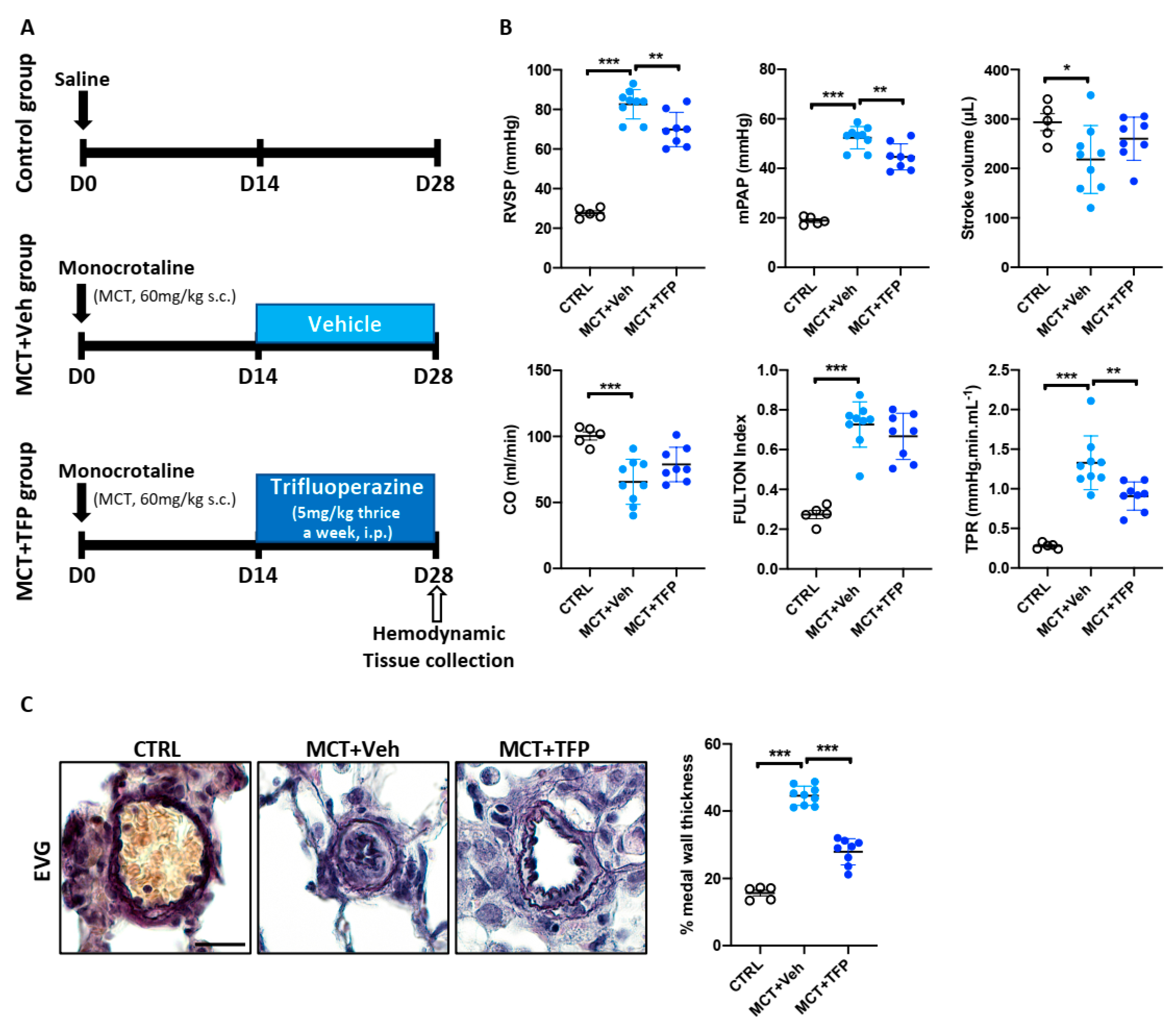
2.6. Beneficial Effects of Trifluoperazine in the Monocrotaline (MCT) PAH Rat Model
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Human Pulmonary Arterial Smooth Muscle Cells
4.2. Cell Culture and Treatments
4.3. Proliferation and Apoptosis Assay
4.4. Western Blotting
4.5. Animal Models
4.6. Hemodynamic Measures of RV Function and Assessment of RV Hypertrophy and PA Wall Thickness
4.7. Immunohistochemistry
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Prisco, S.Z.; Thenappan, T.; Prins, K.W. Treatment Targets for Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2020, 5, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, A.C.; Lauziere, G.; Lega, J.C.; Lacasse, Y.; Martin, S.; Simard, S.; Bonnet, S.; Provencher, S. Combination therapy versus monotherapy for pulmonary arterial hypertension: A meta-analysis. Lancet Respir. Med. 2016, 4, 291–305. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. Translational Advances in the Field of Pulmonary Hypertension. From Cancer Biology to New Pulmonary Arterial Hypertension Therapeutics. Targeting Cell Growth and Proliferation Signaling Hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Garat, C.V.; Crossno, J.T., Jr.; Sullivan, T.M.; Reusch, J.E.; Klemm, D.J. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J. Cardiovasc. Pharmacol. 2013, 62, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Goncharova, E.A.; Ammit, A.J.; Irani, C.; Carroll, R.G.; Eszterhas, A.J.; Panettieri, R.A.; Krymskaya, V.P. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L354–L363. [Google Scholar] [CrossRef] [Green Version]
- Grinnan, D.; Trankle, C.; Andruska, A.; Bloom, B.; Spiekerkoetter, E. Drug repositioning in pulmonary arterial hypertension: Challenges and opportunities. Pulm. Circ. 2019, 9, 2045894019832226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Wu, C.H.; Bai, L.Y.; Tsai, M.H.; Chu, P.C.; Chiu, C.F.; Chen, M.Y.; Chiu, S.J.; Chiang, J.H.; Weng, J.R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents-A drug repurposing strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Chung, Y.M.; Ma, J.; Yang, Q.; Berek, J.S.; Hu, M.C. Pharmacological activation of FOXO3 suppresses triple-negative breast cancer in vitro and in vivo. Oncotarget 2016, 7, 42110–42125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Jia, C.; Xue, Q.; Jiang, J.; Xie, Y.; Wang, R.; Ran, Z.; Xu, F.; Zhang, Y.; Ye, T. Antipsychotic Drug Trifluoperazine Suppresses Colorectal Cancer by Inducing G0/G1 Arrest and Apoptosis. Front. Pharmacol. 2019, 10, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Lan, W.; Fraunhoffer, N.; Meilerman, A.; Iovanna, J.; Santofimia-Castano, P. Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandonselaar, M.; Hickie, R.A.; Quail, J.W.; Delbaere, L.T. Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat. Struct. Biol. 1994, 1, 795–801. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, F.; Xiong, M.; Yang, H.; Lin, W.; Xie, Y.; Xi, H.; Xue, Q.; Ye, T.; Yu, L. Repurposing of antipsychotic trifluoperazine for treating brain metastasis, lung metastasis and bone metastasis of melanoma by disrupting autophagy flux. Pharmacol. Res. 2021, 163, 105295. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, R.; Zhang, C.; Xu, Y.; Han, M.; Huang, B.; Chen, A.; Qiu, C.; Thorsen, F.; Prestegarden, L.; et al. Trifluoperazine, a novel autophagy inhibitor, increases radiosensitivity in glioblastoma by impairing homologous recombination. J. Exp. Clin. Cancer Res. 2017, 36, 118. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 Accumulation Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Hollerhage, M.; Goebel, J.N.; de Andrade, A.; Hildebrandt, T.; Dolga, A.; Culmsee, C.; Oertel, W.H.; Hengerer, B.; Hoglinger, G.U. Trifluoperazine rescues human dopaminergic cells from wild-type alpha-synuclein-induced toxicity. Neurobiol. Aging 2014, 35, 1700–1711. [Google Scholar] [CrossRef]
- Li, A.; Chen, X.; Jing, Z.; Chen, J. Trifluoperazine induces cellular apoptosis by inhibiting autophagy and targeting NUPR1 in multiple myeloma. FEBS Open Bio 2020, 10, 2097–2106. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Furgeson, S.B.; Simpson, P.A.; Park, I.; Vanputten, V.; Horita, H.; Kontos, C.D.; Nemenoff, R.A.; Weiser-Evans, M.C. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc. Res. 2010, 86, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Mitra, A.K.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 induces phosphorylation of PI3K-Akt/PKB to potentiate proliferation of smooth muscle cells in human saphenous vein. Exp. Mol. Pathol. 2010, 89, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savai, R.; Al-Tamari, H.M.; Sedding, D.; Kojonazarov, B.; Muecke, C.; Teske, R.; Capecchi, M.R.; Weissmann, N.; Grimminger, F.; Seeger, W.; et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat. Med. 2014, 20, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Xia, Y.; Gao, T.; Xu, F.; Lei, Q.; Peng, C.; Yang, Y.; Xue, Q.; Hu, X.; Wang, Q.; et al. The antipsychotic agent trifluoperazine hydrochloride suppresses triple-negative breast cancer tumor growth and brain metastasis by inducing G0/G1 arrest and apoptosis. Cell Death Dis. 2018, 9, 1006. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nguyen, D.T.; Olzomer, E.M.; Poon, G.P.; Cole, N.J.; Puvanendran, A.; Phillips, B.R.; Hesselson, D. Rescue of Pink1 Deficiency by Stress-Dependent Activation of Autophagy. Cell Chem. Biol. 2017, 24, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Sun, L.; Zhou, G.; Ge, H.; Meng, Y.; Li, J.; Li, X.; Fang, X. Trifluoperazine as an alternative strategy for the inhibition of tumor growth of colorectal cancer. J. Cell. Biochem. 2019, 120, 15756–15765. [Google Scholar] [CrossRef]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Polischouk, A.G.; Holgersson, A.; Zong, D.; Stenerlow, B.; Karlsson, H.L.; Moller, L.; Viktorsson, K.; Lewensohn, R. The antipsychotic drug trifluoperazine inhibits DNA repair and sensitizes non small cell lung carcinoma cells to DNA double-strand break induced cell death. Mol. Cancer Ther. 2007, 6, 2303–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangopadhyay, S.; Karmakar, P.; Dasgupta, U.; Chakraborty, A. Trifluoperazine stimulates ionizing radiation induced cell killing through inhibition of DNA repair. Mutat. Res. 2007, 633, 117–125. [Google Scholar] [CrossRef]
- Bourgeois, A.; Bonnet, S.; Breuils-Bonnet, S.; Habbout, K.; Paradis, R.; Tremblay, E.; Lampron, M.C.; Orcholski, M.E.; Potus, F.; Bertero, T.; et al. Inhibition of CHK 1 (Checkpoint Kinase 1) Elicits Therapeutic Effects in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Otani, H.; Engelman, R.M.; Rousou, J.A.; Breyer, R.H.; Clement, R.; Prasad, R.; Klar, J.; Das, D.K. Improvement of myocardial function by trifluoperazine, a calmodulin antagonist, after acute coronary artery occlusion and coronary revascularization. J. Thorac. Cardiovasc. Surg. 1989, 97, 267–274. [Google Scholar] [CrossRef]
- Bourgeois, A.; Lambert, C.; Habbout, K.; Ranchoux, B.; Paquet-Marceau, S.; Trinh, I.; Breuils-Bonnet, S.; Paradis, R.; Nadeau, V.; Paulin, R.; et al. FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J. Mol. Med. 2018, 96, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Provencher, S.; Guignabert, C.; Perros, F.; Boucherat, O.; Schermuly, R.T.; Hassoun, P.M.; Rabinovitch, M.; Nicolls, M.R.; Humbert, M. Translating Research into Improved Patient Care in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Omura, J.; Habbout, K.; Shimauchi, T.; Wu, W.H.; Breuils-Bonnet, S.; Tremblay, E.; Martineau, S.; Nadeau, V.; Gagnon, K.; Mazoyer, F.; et al. Identification of Long Noncoding RNA H19 as a New Biomarker and Therapeutic Target in Right Ventricular Failure in Pulmonary Arterial Hypertension. Circulation 2020, 142, 1464–1484. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grobs, Y.; Awada, C.; Lemay, S.-E.; Romanet, C.; Bourgeois, A.; Toro, V.; Nadeau, V.; Shimauchi, K.; Orcholski, M.; Breuils-Bonnet, S.; et al. Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 2919. https://doi.org/10.3390/ijms22062919
Grobs Y, Awada C, Lemay S-E, Romanet C, Bourgeois A, Toro V, Nadeau V, Shimauchi K, Orcholski M, Breuils-Bonnet S, et al. Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2021; 22(6):2919. https://doi.org/10.3390/ijms22062919
Chicago/Turabian StyleGrobs, Yann, Charifa Awada, Sarah-Eve Lemay, Charlotte Romanet, Alice Bourgeois, Victoria Toro, Valérie Nadeau, Kana Shimauchi, Mark Orcholski, Sandra Breuils-Bonnet, and et al. 2021. "Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 22, no. 6: 2919. https://doi.org/10.3390/ijms22062919
APA StyleGrobs, Y., Awada, C., Lemay, S. -E., Romanet, C., Bourgeois, A., Toro, V., Nadeau, V., Shimauchi, K., Orcholski, M., Breuils-Bonnet, S., Tremblay, E., Provencher, S., Paulin, R., Boucherat, O., & Bonnet, S. (2021). Preclinical Investigation of Trifluoperazine as a Novel Therapeutic Agent for the Treatment of Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 22(6), 2919. https://doi.org/10.3390/ijms22062919