
Dopant-Dependent Toxicity of CeO2 Nanoparticles Is Associated with Dynamic Changes in H3K4me3 and H3K27me3 and Transcriptional Activation of NRF2 Gene in HaCaT Human Keratinocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
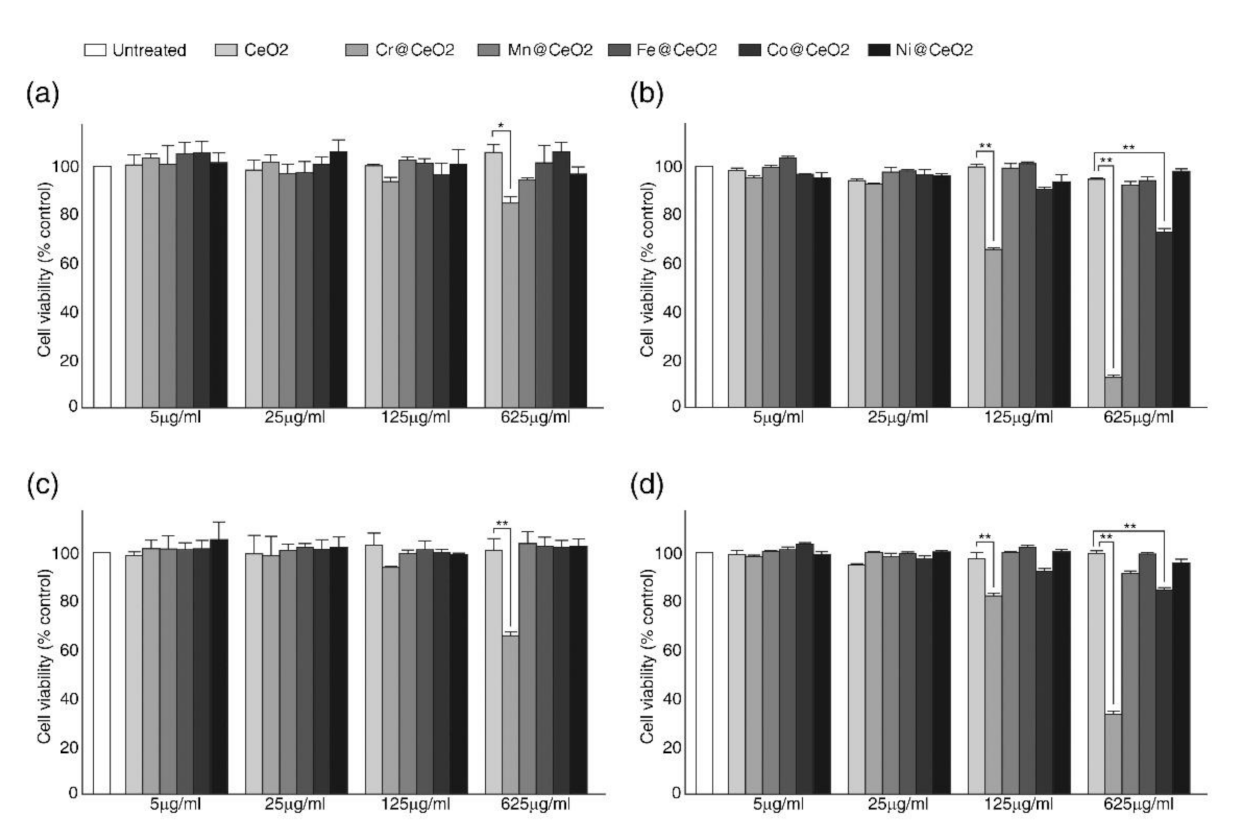
2.1. Effects of Transition Metal Doping on Cell Viability
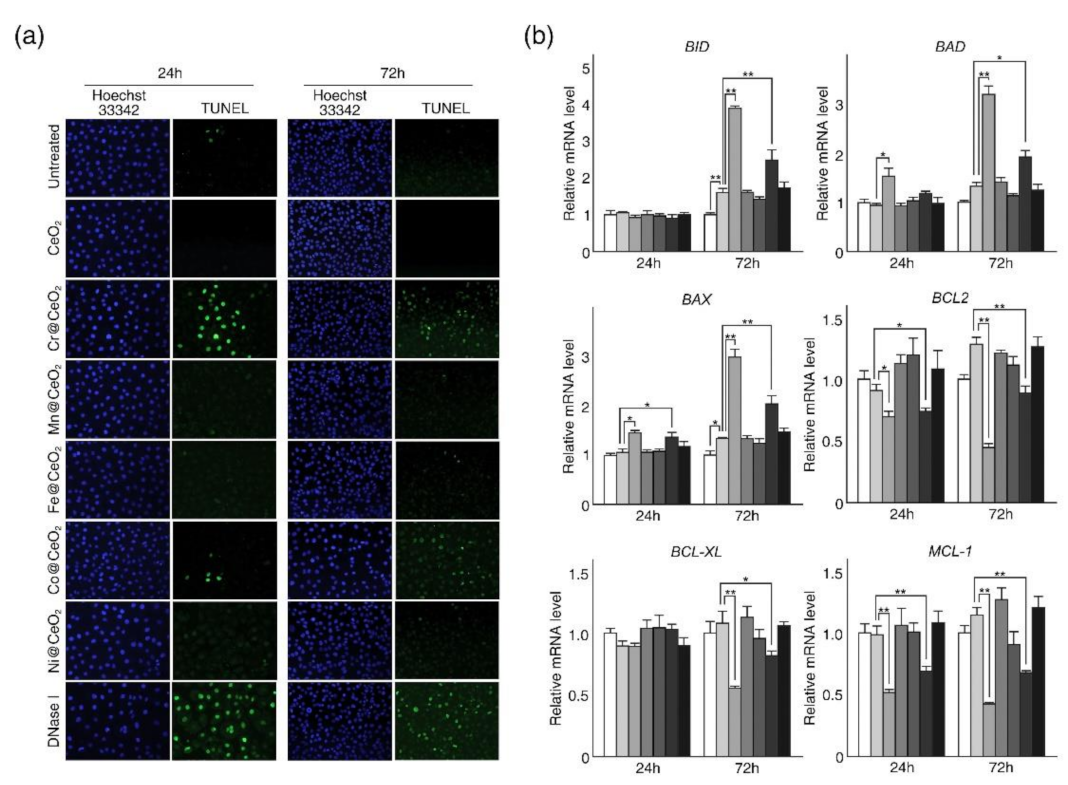
2.2. Dopant-Dependent Toxicities of TM@CeO2 NPs Were Associated with Apoptotic Cell Death in HaCaT Cells
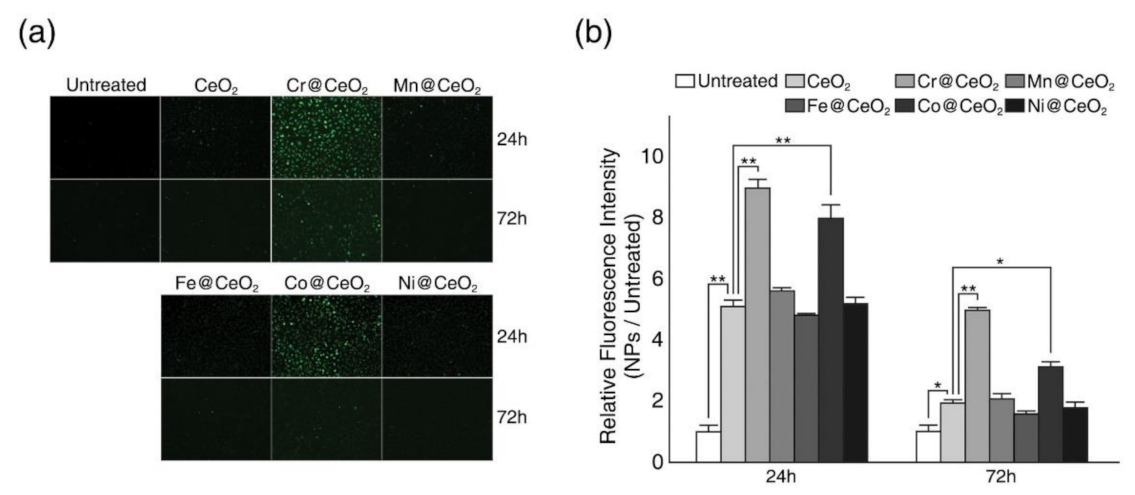
2.3. Effect of Transition Metal Doping on Intracellular ROS Generation
2.4. Dopant-Dependent Toxicity Was Associated with a Failure of Cells to Activate NRF2 Expression
2.5. Dopant Dependent Toxicity Was Associated with H3K4me3 and H3K27me3 Modification at NRF2 Promoter
3. Materials and Methods
3.1. Preparation of Transition Metal-Doped CeO2 NPs
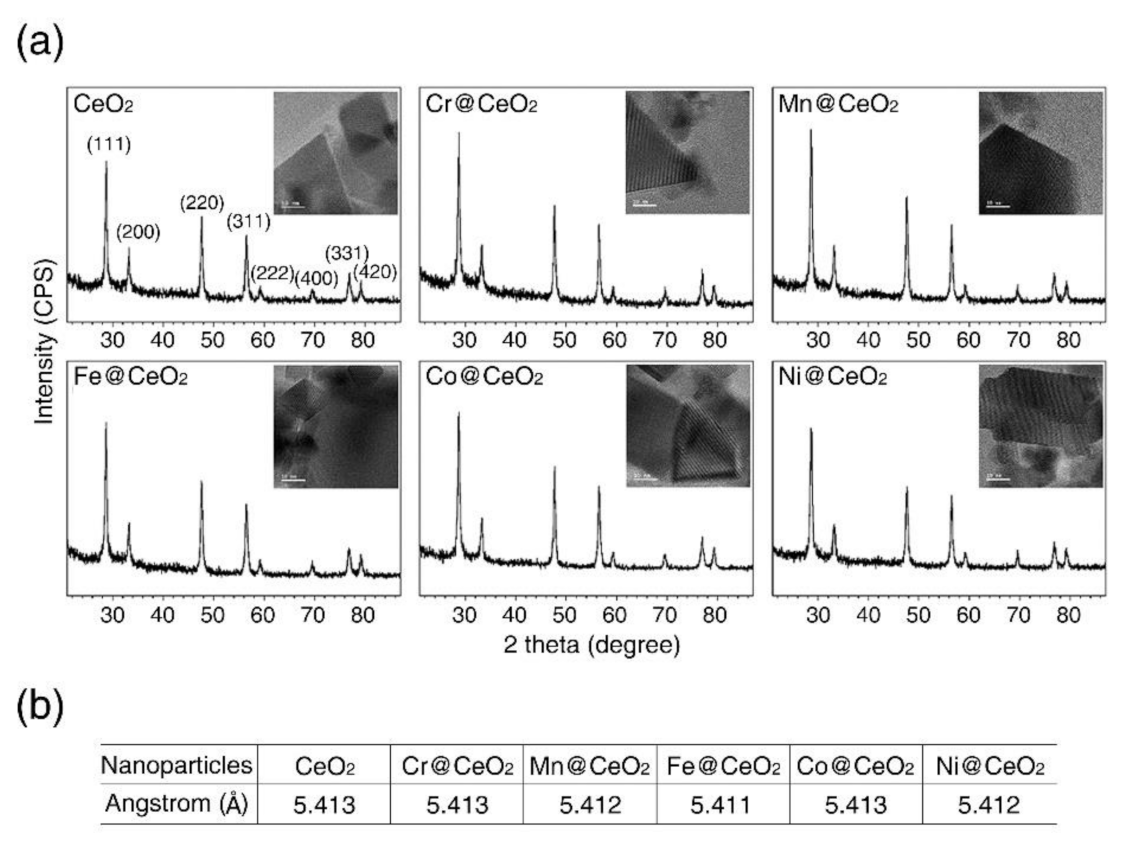
3.2. Characterization of TM@CeO2 NPs
3.3. Cell Culture and NPs Exposure
3.4. Cell Viability Assays
3.5. TUNEL Assay
3.6. Measurement of Intracellular Reactive Oxygen Species (ROS) Levels
3.7. Cell Fractionation and Immunoblot Analysis
3.8. Quantitative RT-PCR Analysis (RT-qPCR)
3.9. Chromatin Immunoprecipitation (ChIP)–qPCR Analysis
3.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, W.J.; Stoessel, P.R.; Wohlleben, W.; Hafner, A. Industrial applications of nanoparticles. Chem. Soc. Rev. 2015, 44, 5793–5805. [Google Scholar] [CrossRef] [Green Version]
- Pelaz, B.; Alexiou, C.; Alvarez-Puebla, R.A.; Alves, F.; Andrews, A.M.; Ashraf, S.; Balogh, L.P.; Ballerini, L.; Bestetti, A.; Brendel, C.; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, K.; Tofail, S.A.M. Nanoparticles in biomedical applications. Adv. Phys. X 2017, 2, 54–88. [Google Scholar] [CrossRef]
- Chavali, M.S.; Nikolova, M.P. Metal oxide nanoparticles and their applications in nanotechnology. SN Appl. Sci. 2019, 1, 607. [Google Scholar] [CrossRef] [Green Version]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djurišić, A.B.; Leung, Y.H.; Ng, A.M.; Xu, X.Y.; Lee, P.K.; Degger, N.; Wu, R.S. Toxicity of metal oxide nanoparticles: Mechanisms, characterization, and avoiding experimental artefacts. Small 2015, 11, 26–44. [Google Scholar] [CrossRef]
- Sengui, A.B.; Asmatulu, E. Toxicity of metal and metal oxide nanoparticles: A review. Environ. Chem. Lett. 2020, 18, 1659–1683. [Google Scholar] [CrossRef]
- Najahi-Missaoui, W.; Arnold, R.D.; Cummings, B.S. Safe Nanoparticles: Are We There Yet? Int. J. Mol. Sci. 2021, 22, 385. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, S.; Lee, J.H.; Yang, J.; Seok, J.; Jung, K.; Lee, J.K. Flow cytometric evaluation of the potential of metal oxide nanoparticles for skin sensitization using 5‑Bromo‑2‑deoxyuridine. Toxicol. Res. 2021. [Google Scholar] [CrossRef]
- Perez, J.M.; Asati, A.; Nath, S.; Kaittanis, C. Synthesis of biocompatible dextran-coated nanoceria with pH-dependent antioxidant properties. Small 2008, 4, 552–556. [Google Scholar] [CrossRef]
- Xia, T.; Kovochich, M.; Liong, M.; Mädler, L.; Gilbert, B.; Shi, H.; Yeh, J.I.; Zink, J.I.; Nel, A.E. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano 2008, 2, 2121–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jiang, H.; Selke, M.; Wang, X. Selective cytotoxicity effect of cerium oxide nanoparticles under UV irradiation. J. Biomed. Nanotechnol. 2014, 10, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, G.; Xu, S.; Chen, S.; Xu, A.; Wu, L. Effect of ionic strength on bioaccumulation and toxicity of silver nanoparticles in Caenorhabditis elegans. Ecotoxicol. Environ. Saf. 2018, 165, 291–298. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, V.; Rizzello, L.; Ingrosso, C.; Liatsi-Douvitsa, E.; De Giorgi, M.L.; De Matteis, G.; Rinaldi, R. Cultivar-dependent anticancer and antibacterial properties of silver nanoparticles synthesized using leaves of different Olea Europaea trees. Nanomaterials 2019, 9, 1544. [Google Scholar] [CrossRef] [Green Version]
- Manke, A.; Wang, L.; Rojanasakul, Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed. Res. Int. 2013, 2013, 942916. [Google Scholar] [CrossRef] [Green Version]
- Pogribna, M.; Hammons, G. Epigenetic Effects of Nanomaterials and Nanoparticles. J. Nanobiotechnol. 2021, 19, 2. [Google Scholar] [CrossRef]
- Liang, F.; Yu, Y.; Zhou, W.; Xu, X.; Zhu, Z. Highly defective CeO2 as a promoter for efficient and stable water oxidation. J. Mater. Chem. A 2015, 3, 634–640. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Mudiyanselage, K.; Al-Shankiti, I.; Foulis, A.; Llorca, J.; Idriss, H. Reactions of ethanol over CeO2 and Ru/CeO2 catalysts. Appl. Catal. B Environ. 2016, 197, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Korsvik, C.; Patil, S.; Seal, S.; Self, W.T. Superoxide dismutase mimetic properties exhibited by vacancy engineered ceria nanoparticles. Chem. Commun. 2007, 10, 1056–1058. [Google Scholar] [CrossRef]
- Asati, A.; Kaittanis, C.; Santra, S.; Perez, J.M. pH-tunable oxidase-like activity of cerium oxide nanoparticles achieving sensitive fluorigenic detection of cancer biomarkers at neutral pH. Anal. Chem. 2011, 83, 2547–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, N.; Manna, P.; Das, J. Synthesis and biomedical applications of nanoceria, a redox active nanoparticle. J. Nanobiotechnol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Liao, X.H.; Zhu, J.M.; Zhu, J.J.; Xu, J.Z.; Chen, H.Y. Preparation of monodispersed nanocrystalline CeO2 powders by microwave irradiation. Chem. Commun. 2001, 10, 937–938. [Google Scholar] [CrossRef]
- Tarnuzzer, R.W.; Colon, J.; Patil, S.; Seal, S. Vacancy engineered ceria nanostructures for protection from radiation-induced cellular damage. Nano Lett. 2005, 5, 2573–2577. [Google Scholar] [CrossRef]
- Goharshadi, E.K.; Samiee, S.; Nancarrow, P. Fabrication of cerium oxide nanoparticles: Characterization and optical properties. J. Colloid Interface Sci. 2011, 356, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhong, L.; Zhong, Q.; Cai, W. Controllable synthesis of CeO2 nanoparticles with different sizes and shapes and their application in NO oxidation. RSC Adv. 2016, 6, 50680–50687. [Google Scholar] [CrossRef]
- Sahoo, T.R.; Armandi, M.; Arletti, R.; Piumetti, M.; Bensaid, S.; Manzoli, M.; Panda, S.R.; Bonelli, B. Pure and Fe-doped CeO2 nanoparticles obtained by microwave assisted combustion synthesis: Physico-chemical properties ruling their catalytic activity towards CO oxidation and soot combustion. Appl. Catal. B Environ. 2017, 211, 31–45. [Google Scholar] [CrossRef]
- Kang, K.; Guo, H.; Varma, A. Noble-metal-free NiCu/CeO2 catalysts for H2 generation from hydrous hydrazine. Appl. Catal. B Environ. 2019, 249, 54–62. [Google Scholar] [CrossRef]
- Choi, J.H.; Hong, J.A.; Son, Y.R.; Wang, J.; Kim, H.S.; Lee, H.; Lee, H. Comparison of Enhanced Photocatalytic Degradation Efficiency and Toxicity Evaluations of CeO2 Nanoparticles Synthesized Through Double-Modulation. Nanomaterials 2020, 10, 1543. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, Y.W.; Zhou, X.D.; Ma, Y. Toxicity of cerium oxide nanoparticles in human lung cancer cells. Int. J. Toxicol. 2006, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Thill, A.; Zeyons, O.; Spalla, O.; Chauvat, F.; Rose, J.; Auffan, M.; Flank, A.M. Cytotoxicity of CeO2 nanoparticles for Escherichia coli. Physico-chemical insight of the cytotoxicity mechanism. Environ. Sci. Technol. 2006, 40, 6151–6156. [Google Scholar] [CrossRef] [PubMed]
- Rodea-Palomares, I.; Boltes, K.; Fernández-Piñas, F.; Leganés, F.; García-Calvo, E.; Santiago, J.; Rosal, R. Physicochemical characterization and ecotoxicological assessment of CeO2 nanoparticles using two aquatic microorganisms. Toxicol. Sci. 2011, 119, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Patil, S.; Seal, S.; McGinnis, J.F. Rare earth nanoparticles prevent retinal degeneration induced by intracellular peroxides. Nat. Nanotechnol. 2006, 1, 142–150. [Google Scholar] [CrossRef]
- Niu, J.; Azfer, A.; Rogers, L.M.; Wang, X.; Kolattukudy, P.E. Cardioprotective effects of cerium oxide nanoparticles in a transgenic murine model of cardiomyopathy. Cardiovasc. Res. 2007, 73, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.K.; Kim, T.; Choi, I.Y.; Soh, M.; Kim, D.; Kim, Y.-J.; Jang, H.; Yang, H.-S.; Kim, J.Y.; Park, H.-K.; et al. Ceria nanoparticles that can protect against ischemic stroke. Angew. Chem. Int. Ed. 2012, 51, 11039–11043. [Google Scholar] [CrossRef]
- Park, E.J.; Choi, J.; Park, Y.K.; Park, K. Oxidative stress induced by cerium oxide nanoparticles in cultured BEAS-2B cells. Toxicology 2008, 245, 90–100. [Google Scholar] [CrossRef]
- Auffan, M.; Rose, J.; Orsiere, T.; De Meo, M.; Thill, A.; Zeyons, O.; Proux, O.; Masion, A.; Chaurand, P.; Spalla, O.; et al. CeO2 nanoparticles induce DNA damage towards human dermal fibroblasts in vitro. Nanotoxicology 2009, 3, 161–171. [Google Scholar] [CrossRef]
- Eom, H.J.; Choi, J. Oxidative stress of CeO2 nanoparticles via p38-Nrf-2 signaling pathway in human bronchial epithelial cell, Beas-2B. Toxicol. Lett. 2009, 187, 77–83. [Google Scholar] [CrossRef]
- Hussain, S.; Al-Nsour, F.; Rice, A.B.; Marshburn, J.; Yingling, B.; Ji, Z.; Zink, J.I.; Walker, N.J.; Garantziotis, S. Cerium dioxide nanoparticles induce apoptosis and autophagy in human peripheral blood monocytes. ACS Nano 2012, 6, 5820–5829. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Itoha, K.; Chibabc, T.; Takahashia, S.; Ishiia, T.; Igarashia, K.; Katoha, Y.; Oyaked, T.; Hayashid, N.; Satohe, K.; Hatayamae, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Wu, R.; Guo, Y.; Kong, A.N. Regulation of Keap1-Nrf2 signaling: The role of epigenetics. Curr. Opin. Toxicol. 2016, 1, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanvand, D.; Amiri, I.; Asl, S.S.; Saidijam, M.; Shabab, N.; Artimani, T. Effects of CeO2 nanoparticles on the HO-1, NQO1, and GCLC expression in the testes of diabetic rats. Can. J. Physiol. Pharmacol. 2018, 96, 963–969. [Google Scholar] [CrossRef]
- Artimani, T.; Amiri, I.; Asl, S.S.; Saidijam, M.; Hasanvand, D.; Afshar, S. Amelioration of diabetes-induced testicular and sperm damage in rats by cerium oxide nanoparticle treatment. Andrologia 2018, 50, e13089. [Google Scholar] [CrossRef]
- Prasad, R.Y.; McGee, J.K.; Killius, M.G.; Suarez, D.A.; Blackman, C.F.; De Marini, D.M.; Simmons, S.O. Investigating oxidative stress and inflammatory responses elicited by silver nanoparticles using high-throughput reporter genes in HepG2 cells: Effect of size, surface coating, and intracellular uptake. Toxicol. Vitro 2013, 27, 2013–2021. [Google Scholar] [CrossRef]
- Hashem, R.M.; Rashd, L.A.; Hashem, K.S.; Soliman, H.M. Cerium oxide nanoparticles alleviate oxidative stress and decreases Nrf-2/HO-1 in D-GALN/LPS induced hepatotoxicity. Biomed. Pharmacother. 2015, 73, 80–86. [Google Scholar] [CrossRef]
- Singh, R.; Karakoti, A.S.; Self, W.; Seal, S.; Singh, S. Redox-Sensitive Cerium Oxide Nanoparticles Protect Human Keratinocytes from Oxidative Stress Induced by Glutathione Depletion. Langmuir 2016, 32, 12202–12211. [Google Scholar] [CrossRef]
- Li, Y.; Hou, X.; Yang, C.; Pang, Y.; Li, X.; Jiang, G.; Liu, Y. Photoprotection of Cerium Oxide Nanoparticles against UVA radiation-induced Senescence of Human Skin Fibroblasts due to their Antioxidant Properties. Sci. Rep. 2019, 9, 2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Van de Loosdrecht, A.A.; Nennie, E.; Ossenkoppele, G.J.; Beelen, R.H.; Langenhuijsen, M.M. Cell mediated cytotoxicity against U 937 cells by human monocytes and macrophages in a modified colorimetric MTT assay. A methodological study. J. Immunol. Methods 1991, 141, 15–22. [Google Scholar] [CrossRef]
- Repetto, G.; Del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoek, T.L.; Li, C.; Shao, Z.; Schumacker, P.T.; Becker, L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997, 29, 2571–2583. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lee, H. Histone demethylase KDM4D cooperates with NFIB and MLL1 complex to regulate adipogenic differentiation of C3H10T1/2 mesenchymal stem cells. Sci. Rep. 2020, 10, 3050. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Lee, H.; Lee, H.; Lee, H. Dopant-Dependent Toxicity of CeO2 Nanoparticles Is Associated with Dynamic Changes in H3K4me3 and H3K27me3 and Transcriptional Activation of NRF2 Gene in HaCaT Human Keratinocytes. Int. J. Mol. Sci. 2021, 22, 3087. https://doi.org/10.3390/ijms22063087
Choi JH, Lee H, Lee H, Lee H. Dopant-Dependent Toxicity of CeO2 Nanoparticles Is Associated with Dynamic Changes in H3K4me3 and H3K27me3 and Transcriptional Activation of NRF2 Gene in HaCaT Human Keratinocytes. International Journal of Molecular Sciences. 2021; 22(6):3087. https://doi.org/10.3390/ijms22063087
Chicago/Turabian StyleChoi, Jang Hyun, Haram Lee, Hangil Lee, and Hansol Lee. 2021. "Dopant-Dependent Toxicity of CeO2 Nanoparticles Is Associated with Dynamic Changes in H3K4me3 and H3K27me3 and Transcriptional Activation of NRF2 Gene in HaCaT Human Keratinocytes" International Journal of Molecular Sciences 22, no. 6: 3087. https://doi.org/10.3390/ijms22063087
APA StyleChoi, J. H., Lee, H., Lee, H., & Lee, H. (2021). Dopant-Dependent Toxicity of CeO2 Nanoparticles Is Associated with Dynamic Changes in H3K4me3 and H3K27me3 and Transcriptional Activation of NRF2 Gene in HaCaT Human Keratinocytes. International Journal of Molecular Sciences, 22(6), 3087. https://doi.org/10.3390/ijms22063087