
Role of the Innate Immunity Signaling Pathway in the Pathogenesis of Sjögren’s Syndrome



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
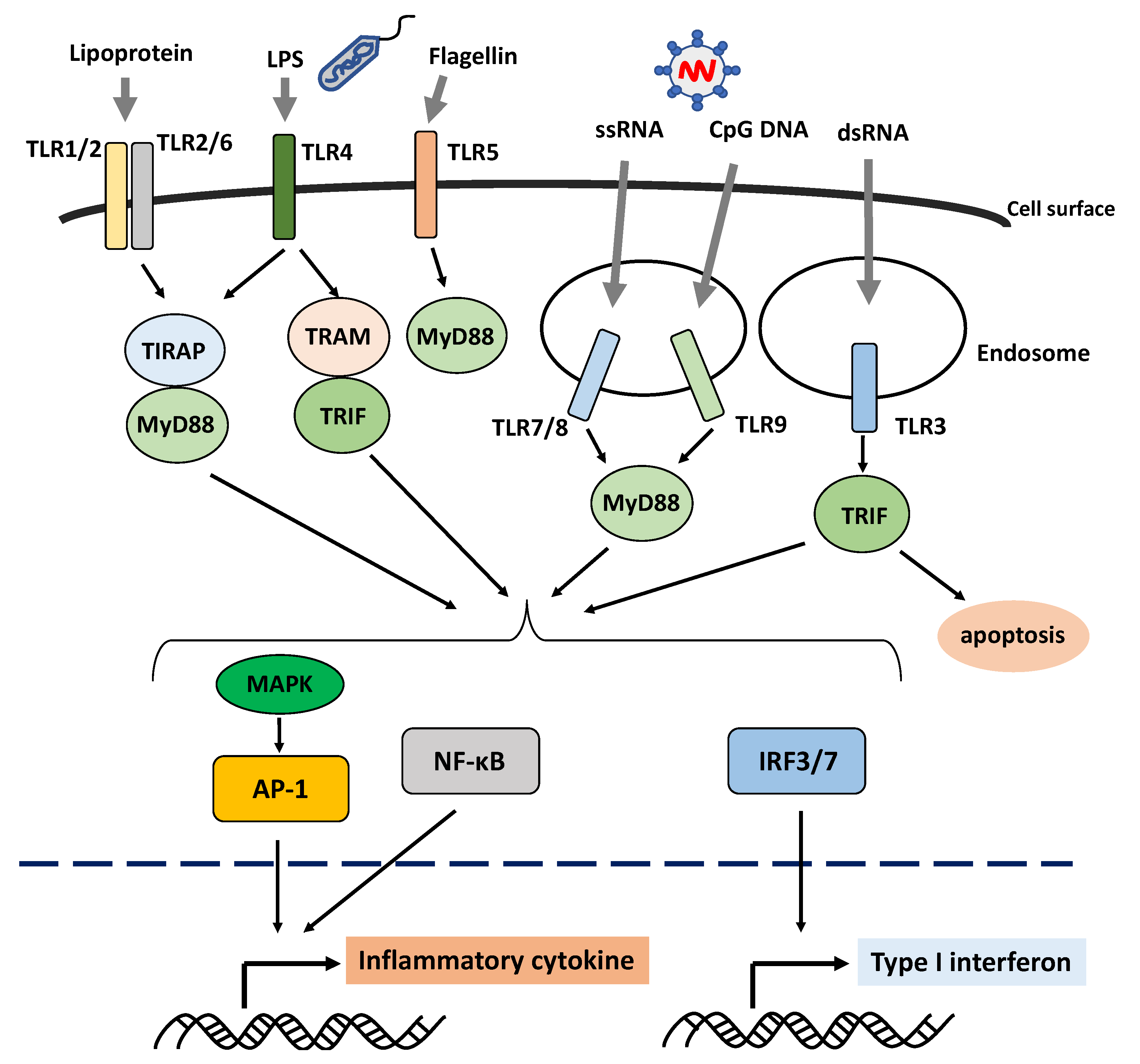
2. Innate Immune Response
3. Role of Cell Surface TLRs in SS
3.1. TLR1, TLR2 and TLR6 in SS
3.2. TLR4 in SS
3.3. TLR5 in SS
4. Role of Endosomal TLRs in SS
4.1. TLR3, TLR7–9 in SS
4.2. TLR7, TLR9, and Sex Differences in SS
5. Role of Cytoplasmic RNA and DNA Sensors in SS
6. Role of Endosomal TLRs in Type I IFN-Mediated B Cell Activation
7. Role of Inflammasome in SS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramos-Casals, M.; Tzioufas, A.G.; Font, J. Primary Sjogren’s syndrome: New clinical and therapeutic concepts. Ann. Rheum. Dis. 2005, 64, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Bournia, V.K.; Vlachoyiannopoulos, P.G. Subgroups of Sjögren syndrome patients according to serological profiles. J. Autoimmun. 2012, 39, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.; Sterin-Borda, L.; Camusso, J.J.; Arana, R.; Hubscher, O.; Borda, E. Circulating antibodies against rat parotid gland M3 muscarinic receptors in primary Sjögren’s syndrome. Clin. Exp. Immunol. 1996, 104, 454–459. [Google Scholar] [CrossRef]
- Haneji, N.; Nakamura, T.; Takio, K.; Yanagi, K.; Higashiyama, H.; Saito, I.; Noji, S.; Sugino, H.; Hayashi, Y. Identification of alpha-fodrin as a candidate autoantigen in primary Sjögren’s syndrome. Science 1997, 276, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, H.; Asashima, H.; Takai, C.; Hagiwara, S.; Hagiya, C.; Yokosawa, M.; Hirota, T.; Umehara, H.; Kawakami, A.; Nakamura, H.; et al. Primary and secondary surveys on epidemiology of Sjögren’s syndrome in Japan. Mod. Rheumatol. 2014, 24, 464–470. [Google Scholar] [CrossRef]
- Kang, H.I.; Fei, H.M.; Saito, I.; Sawada, S.; Chen, S.L.; Yi, D.; Chan, E.; Peebles, C.; Bugawan, T.L.; Erlich, H.A.; et al. Comparison of HLA class II genes in Caucasoid, Chinese, and Japanese patients with primary Sjögren’s syndrome. J. Immunol. 1993, 150, 3615–3623. [Google Scholar]
- Ishimaru, N.; Arakaki, R.; Yoshida, S.; Yamada, A.; Noji, S.; Hayashi, Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren’s syndrome-like autoimmune exocrinopathy. J. Exp. Med. 2008, 205, 2915–2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, I.; Servenius, B.; Compton, T.; Fox, R.I. Detection of Epstein–Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjogren’s syndrome. J. Exp. Med. 1989, 169, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Saito, I.; Shimuta, M.; Terauchi, K.; Tsubota, K.; Yodoi, J.; Miyasaka, N. Increased expression of human thioredoxin/adult T cell leukemia-derived factor in Sjögren’s syndrome. Arthritis Rheum. 1996, 39, 773–782. [Google Scholar] [CrossRef]
- McArthur, C.; Wang, Y.; Veno, P.; Zhang, J.; Fiorella, R. Intracellular trafficking and surface expression of SS-A (Ro), SS-B (La), poly(ADP-ribose) polymerase and alpha-fodrin autoantigens during apoptosis in human salivary gland cells induced by tumour necrosis factor-alpha. Arch. Oral. Biol. 2002, 47, 443–448. [Google Scholar] [CrossRef]
- Naito, Y.; Matsumoto, I.; Wakamatsu, E.; Goto, D.; Ito, S.; Tsutsumi, A.; Sumida, T. Altered peptide ligands regulate muscarinic acetylcholine receptor reactive T cells of patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2006, 65, 269–271. [Google Scholar] [CrossRef]
- Kong, L.; Ogawa, N.; Nakabayashi, T.; Liu, G.T.; D’Souza, E.; McGuff, H.S.; Guerrero, D.; Talal, N.; Dang, H. Fas and Fas ligand expression in the salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 1997, 40, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Koji, T.; Tominaga, M.; Kawakami, A.; Migita, K.; Kawabe, Y.; Nakamura, T.; Shirabe, S.; Eguchi, K. Apoptosis in labial salivary glands from Sjögren’s syndrome (SS) patients: Comparison with human T lymphotropic virus-I (HTLV-I)-seronegative and -seropositive SS patients. Clin. Exp. Immunol. 1998, 114, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kawakami, A.; Izumi, M.; Nakashima, T.; Takagi, Y.; Ida, H.; Nakamura, T.; Nakamura, T.; Eguchi, K. Detection of the soluble form of Fas ligand (sFasL) and sFas in the saliva from patients with Sjögren’s syndrome. Clin. Exp. Rheumatol. 2005, 23, 915. [Google Scholar] [PubMed]
- Tsubota, K.; Saito, I.; Miyasaka, N. Granzyme A and perforin expressed in the lacrimal glands of patients with Sjögren’s syndrome. Am. J. Ophthalmol. 1994, 117, 120–121. [Google Scholar] [CrossRef]
- Fujihara, T.; Fujita, H.; Tsubota, K.; Saito, K.; Tsuzaka, K.; Abe, T.; Takeuchi, T. Preferential localization of CD8+ alpha E beta 7+ T cells around acinar epithelial cells with apoptosis in patients with Sjögren’s syndrome. J. Immunol. 1999, 163, 2226–2235. [Google Scholar] [PubMed]
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef]
- Daniels, T.E.; Cox, D.; Shiboski, C.H.; Schiødt, M.; Wu, A.; Lanfranchi, H.; Umehara, H.; Zhao, Y.; Challacombe, S.; Lam, M.Y.; et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjögren’s syndrome among 1,726 registry participants. Arthritis Rheum. 2011, 63, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef]
- Sumida, T.; Azuma, N.; Moriyama, M.; Takahashi, H.; Asashima, H.; Honda, F.; Abe, S.; Ono, Y.; Hirota, T.; Hirata, S.; et al. Clinical practice guideline for Sjögren’s syndrome 2017. Mod. Rheumatol. 2018, 28, 383–408. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Pattern recognition theory and the launch of modern innate immunity. J. Immunol. 2013, 191, 4473–4474. [Google Scholar] [CrossRef]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorov, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Reksten, T.R.; Ice, J.A.; Kelly, J.A.; Adrianto, I.; Rasmussen, A.; Wang, S.; He, B.; Grundahl, K.M.; Glenn, S.B.; et al. Identification of a Sjogren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet. 2017, 13, e1006820. [Google Scholar] [CrossRef] [Green Version]
- Emamian, E.S.; Leon, J.M.; Lessard, C.J.; Grandits, M.; Baechler, E.C.; Gaffney, P.M.; Segal, B.; Rhodus, N.L.; Moser, K.L. Peripheral blood gene expression profiling in Sjögren’s syndrome. Genes Immun. 2009, 10, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Hjelmervik, T.O.; Petersen, K.; Jonassen, I.; Jonsson, R.; Bolstad, A.I. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum. 2005, 52, 1534–1544. [Google Scholar] [CrossRef]
- Kimoto, O.; Sawada, J.; Shimoyama, K.; Suzuki, D.; Nakamura, S.; Hayashi, H.; Ogawa, N. Activation of the interferon pathway in peripheral blood of patients with Sjogren’s syndrome. J. Rheumatol. 2011, 38, 310–316. [Google Scholar] [CrossRef]
- Vakaloglou, K.M.; Mavragani, C.P. Activation of the type I interferon pathway in primary Sjogren’s syndrome: An update. Curr. Opin. Rheumatol. 2011, 23, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Strauß, R.; Rose, T.; Flint, S.M.; Klotsche, J.; Häupl, T.; Peck-Radosavljevic, M.; Yoshida, T.; Kyogoku, C.; Flechsig, A.; Becker, A.M.; et al. Type I interferon as a biomarker in autoimmunity and viral infection: A leukocyte subset-specific analysis unveils hidden diagnostic options. J. Mol. Med. 2017, 95, 753–765. [Google Scholar] [CrossRef]
- Bodewes, I.L.A.; Al-Ali, S.; van Helden-Meeuwsen, C.G.; Maria, N.I.; Tarn, J.; Lendrem, D.W.; Schreurs, M.W.J.; Steenwijk, E.C.; van Daele, P.L.A.; Both, T.; et al. Systemic interferon type I and type II signatures in primary Sjögren’s syndrome reveal differences in biological disease activity. Rheumatology 2018, 57, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of intrinsic epithelial activation in the pathogenesis of Sjogren’s syndrome. J. Autoimmun. 2010, 35, 219–224. [Google Scholar] [CrossRef]
- Dimitriou, I.D.; Kapsogeorgou, E.K.; Abu-Helu, R.F.; Moutsopoulos, H.M.; Manoussakis, M.N. Establishment of a convenient system for the long-term culture and study of non-neoplastic human salivary gland epithelial cells. Eur. J. Oral. Sci. 2002, 110, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Ping, L.; Zhenjun, L.; Takada, Y.; Sugai, S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjögren’s syndrome. Arthritis Rheum. 2002, 46, 2730–2741. [Google Scholar] [CrossRef]
- Shimizu, S.; Kurashige, Y.; Nishimura, M.; Yamazaki, M.; Sato, J.; Saitoh, M.; Selimovic, D.; Abiko, Y. Involvement of toll-like receptors in autoimmune sialoadenitis of the non-obese diabetic mouse. J. Oral. Pathol. Med. 2012, 41, 517–523. [Google Scholar] [CrossRef]
- Kiripolsky, J.; McCabe, L.G.; Gaile, D.P.; Kramer, J.M. Myd88 is required for disease development in a primary Sjögren’s syndrome mouse model. J. Leukoc. Biol. 2017, 102, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.S.; Wee, Y.; Yang, C.H.; Melvin, J.E.; Baker, O.J. ALX/FPR2 modulates anti-inflammatory responses in mouse submandibular gland. Sci. Rep. 2016, 6, 24244. [Google Scholar] [CrossRef]
- Kwok, S.K.; Cho, M.L.; Her, Y.M.; Oh, H.J.; Park, M.K.; Lee, S.Y.; Woo, Y.J.; Ju, J.H.; Park, K.S.; Kim, H.Y.; et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res. Ther. 2012, 14, R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Patidar, M.; Yadav, N.; Dalai, S.K. Interleukin 15: A key cytokine for immunotherapy. Cytokine Growth Factor Rev. 2016, 31, 49–59. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Lisi, S. Interleukin-15 as a potential new target in Sjögren’s syndrome-associated inflammation. Pathology 2016, 48, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, A.; Nakashima, K.; Tamai, M.; Nakamura, H.; Iwanaga, N.; Fujikawa, K.; Aramaki, T.; Arima, K.; Iwamoto, N.; Ichinose, K.; et al. Toll-like receptor in salivary glands from patients with Sjogren’s syndrome: Functional analysis by human salivary gland cell line. J. Rheumatol. 2007, 34, 1019–1026. [Google Scholar] [PubMed]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjögren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef]
- Barrera, M.J.; Aguilera, S.; Veerman, E.; Quest, A.F.; Diaz-Jimenez, D.; Urzua, U.; Cortés, J.; González, S.; Castro, I.; Molina, C.; et al. Salivary mucins induce a toll-like receptor 4-mediated pro-inflammatory response in human submandibular salivary cells: Are mucins involved in Sjogren’s syndrome? Rheumatology 2015, 54, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Haruta, I.; Shibata, N.; Yanagisawa, N.; Yagi, J. Flagellar filament structural protein induces Sjögren’s syndrome-like sialadenitis in mice. Oral Dis. 2017, 23, 636–643. [Google Scholar] [CrossRef]
- Karlsen, M.; Jakobsen, K.; Jonsson, R.; Hammenfors, D.; Hansen, T.; Appel, S. Expression of toll-like receptors in peripheral blood mononuclear cells of patients with primary Sjögren’s syndrome. Scand. J. Immunol. 2017, 85, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Duffy, L.; O’Reilly, S.C. Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. Immunotargets Ther. 2016, 5, 69–80. [Google Scholar] [PubMed] [Green Version]
- Brentano, F.; Schorr, O.; Gay, R.E.; Gay, S.; Kyburz, D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum. 2005, 52, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Nagashima, H.; Brown, V.B.; Peck, A.B.; Humphreys-Beher, M.G. Two NOD Idd-associated intervals contribute synergistically to the development of autoimmune exocrinopathy (Sjögren’s syndrome) on a healthy murine background. Arthritis Rheum. 2002, 46, 1390–1398. [Google Scholar] [CrossRef]
- Killedar, S.J.; Eckenrode, S.E.; McIndoe, R.A.; She, J.X.; Nguyen, C.Q.; Peck, A.B.; Cha, S. Early pathogenic events associated with Sjögren’s syndrome (SjS)-like disease of the NOD mouse using microarray analysis. Lab. Investig. 2006, 86, 1243–1260. [Google Scholar] [CrossRef]
- Deshmukh, U.S.; Nandula, S.R.; Thimmalapura, P.R.; Scindia, Y.M.; Bagavant, H. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J. Oral. Pathol. Med. 2009, 38, 42–47. [Google Scholar] [CrossRef]
- Nandula, S.R.; Scindia, Y.M.; Dey, P.; Bagavant, H.; Deshmukh, U.S. Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. Oral. Dis. 2011, 17, 801–807. [Google Scholar] [CrossRef]
- Nandula, S.R.; Dey, P.; Corbin, K.L.; Nunemaker, C.S.; Bagavant, H.; Deshmukh, U.S. Salivary gland hypofunction induced by activation of innate immunity is dependent on type I interferon signaling. J. Oral. Pathol. Med. 2013, 42, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Horai, Y.; Suzuki, T.; Okada, A.; Ichinose, K.; Yamasaki, S.; Koji, T.; Kawakami, A. TLR3-mediated apoptosis and activation of phosphorylated Akt in the salivary gland epithelial cells of primary Sjogren’s syndrome patients. Rheumatol. Int. 2013, 33, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J. Autoimmun. 2010, 35, 212–218. [Google Scholar] [CrossRef]
- Horai, Y.; Nakamura, H.; Nakashima, Y.; Hayashi, T.; Kawakami, A. Analysis of the downstream mediators of toll-like receptor 3-induced apoptosis in labial salivary glands in patients with Sjogren’s syndrome. Mod. Rheumatol. 2016, 26, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin. Exp. Immunol. 2014, 178, 548–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Sellam, J.; Eid, P.; Lebon, P.; Lepajolec, C.; Mariette, X. Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways. Eur. J. Immunol. 2008, 38, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; Steenwijk, E.C.; IJpma, A.S.; van Helden-Meeuwsen, C.G.; Vogelsang, P.; Beumer, W.; Brkic, Z.; van Daele, P.L.; van Hagen, P.M.; van der Spek, P.J.; et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2017, 76, 721–730. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Z.; Yu, C.; Yang, C. Expression of toll-like receptors 7, 8, and 9 in primary Sjogren’s syndrome. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol Endod. 2010, 109, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Nakamura, H.; Takatani, A.; Umeda, M.; Horai, Y.; Kurushima, S.; Michitsuji, T.; Nakashima, Y.; Kawakami, A. Activation of toll-like receptor 7 signaling in labial salivary glands of primary Sjogren’s syndrome patients. Clin. Exp. Immunol. 2019, 196, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.; Sarkar, I.; Hammenfors, D.; Bergum, B.; Vogelsang, P.; Solberg, S.M.; Gavasso, S.; Brun, J.G.; Jonsson, R.; Appel, S. Single cell based phosphorylation profiling identifies alterations in toll-like receptor 7 and 9 signaling in patients with primary Sjögren’s syndrome. Front Immunol. 2019, 10, 281. [Google Scholar] [CrossRef]
- Aota, K.; Yamanoi, T.; Kani, K.; Ono, S.; Momota, Y.; Azuma, M. Inhibition of JAK-STAT Signaling by Baricitinib Reduces Interferon-gamma-Induced CXCL10 Production in Human Salivary Gland Ductal Cells. Inflammation 2021, 44, 206–216. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Kwok, S.K.; Baek, S.; Jang, S.G.; Hong, S.M.; Min, J.W.; Choi, S.S.; Lee, J.; Cho, M.L.; et al. JAK-1 Inhibition Suppresses Interferon-Induced BAFF Production in Human Salivary Gland: Potential Therapeutic Strategy for Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 2057–2066. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Kiripolsky, J.; Klimatcheva, E.; Howell, A.; Fereidouni, F.; Levenson, R.; Rothstein, T.L.; Kramer, J.M. Early BAFF receptor blockade mitigates murine Sjögren’s syndrome: Concomitant targeting of CXCL13 and the BAFF receptor prevents salivary hypofunction. Clin. Immunol. 2016, 164, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Fukui, R.; Saitoh, S.; Matsumoto, F.; Kozuka-Hata, H.; Oyama, M.; Tabeta, K.; Beutler, B.; Miyake, K. Unc93B1 biases toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J. Exp. Med. 2009, 206, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, K.M.; Wang, Y.; Bastacky, S.; Shlomchik, M.J. Toll-like receptor 9 suppresses lupus disease in Fas-sufficient MRL Mice. PLoS ONE 2017, 12, e0173471. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Watanabe, M.; Kobayashi, M.; Miyazaki, K.; Hayashi, Y. Development of autoimmune exocrinopathy resembling Sjögren’s syndrome in estrogen-deficient mice of healthy background. Am. J. Pathol. 2003, 163, 1481–1490. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef]
- Mougeot, J.L.; Noll, B.D.; Bahrani Mougeot, F.K. Sjogren’s syndrome X-chromosome dose effect: An epigenetic perspective. Oral. Dis. 2019, 25, 372–384. [Google Scholar] [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles-mechanism for the loss of immunological tolerance in Sjögren’s syndrome. Clin. Exp. Immunol. 2018, 191, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Wang, J.; Dai, M.; Cui, Y.; Hou, G.; Deng, J.; Gao, X.; Liao, Z.; Liu, Y.; Meng, Y.; Wu, L. Elevated IFIT3 contributes to abnormal overactive cGAS-STING signaling in human systemic lupus erythematosus monocytes. Arthritis Rheumatol. 2018, 70, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, L.; Ambrosi, A.; Espinosa, A.; Ottosson, L.; Eloranta, M.L.; Zhou, W.; Elfving, A.; Greenfield, E.; Kuchroo, V.K.; Wahren-Herlenius, M. Interferon-alpha induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J. Clin. Immunol. 2008, 28, 220–231. [Google Scholar] [CrossRef]
- Karlsen, M.; Hansen, T.; Nordal, H.H.; Brun, J.G.; Jonsson, R.; Appel, S. Expression of toll-like receptor -7 and -9 in B cell subsets from patients with primary Sjogren’s syndrome. PLoS ONE 2015, 10, e0120383. [Google Scholar] [CrossRef] [Green Version]
- Brauner, S.; Folkersen, L.; Kvarnström, M.; Meisgen, S.; Petersen, S.; Franzén-Malmros, M.; Mofors, J.; Brokstad, K.A.; Klareskog, L.; Jonsson, R.; et al. H1N1 vaccination in Sjögren’s syndrome triggers polyclonal B cell activation and promotes autoantibody production. Ann. Rheum. Dis. 2017, 76, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B cell responses through toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, J.; Tluk, S.; Schmitz, C.; Hamm, S.; Jurk, M.; Forsbach, A.; Akira, S.; Kelly, K.M.; Reeves, W.H.; Bauer, S.; et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves toll-like receptors 7 and 8. J. Exp. Med. 2005, 202, 1575–1785. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.H.; Gordon, T.P. Autoimmunity: Ro60-associated RNA takes its toll on disease pathogenesis. Nat. Rev. Rheumatol. 2016, 12, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian-Ding, I.B.; Wagner, M.; Hornung, V.; Giese, T.; Schnurr, M.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J. Immunol. 2005, 174, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, I.L.A.; Bjork, A.; Versnel, M.A.; Wahren-Herlenius, M. Innate immunity and interferons in the pathogenesis of Sjogren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef]
- Batten, M.; Groom, J.; Cachero, T.G.; Qian, F.; Schneider, P.; Tschopp, J.; Browning, J.L.; Mackay, F. BAFF mediates survival of peripheral immature B lymphocytes. J. Exp. Med. 2000, 192, 1453–1466. [Google Scholar] [CrossRef] [Green Version]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Investig. 2003, 112, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Groom, J.; Kalled, S.L.; Cutler, A.H.; Olson, C.; Woodcock, S.A.; Schneider, P.; Tschopp, J.; Cachero, T.G.; Batten, M.; Wheway, J.; et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J. Clin. Investig. 2002, 109, 59–68. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Eric Gottenberg, J.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjogren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef] [Green Version]
- Pollard, R.P.; Abdulahad, W.H.; Vissink, A.; Hamza, N.; Burgerhof, J.G.; Meijer, J.M.; Visser, A.; Huitema, M.G.; Spijkervet, F.K.; Kallenberg, C.G.; et al. Serum levels of BAFF, but not APRIL, are increased after rituximab treatment in patients with primary Sjogren’s syndrome: Data from a placebo-controlled clinical trial. Ann. Rheum. Dis. 2013, 72, 146–148. [Google Scholar] [CrossRef] [Green Version]
- Lavie, F.; Miceli-Richard, C.; Quillard, J.; Roux, S.; Leclerc, P.; Mariette, X. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with Sjögren’s syndrome. J. Pathol. 2004, 202, 496–502. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Kang, I. Advances in disease mechanisms and translational technologies: Clinicopathologic significance of inflammasome activation in autoimmune diseases. Arthritis Rheumatol. 2020, 72, 386–395. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Baldini, C.; Rossi, C.; Ferro, F.; Santini, E.; Seccia, V.; Donati, V.; Solini, A. The P2 × 7 receptor-inflammasome complex has a role in modulating the inflammatory response in primary Sjögren’s syndrome. J. Intern. Med. 2013, 274, 480–489. [Google Scholar] [CrossRef]
- Woods, L.T.; Camden, J.M.; Batek, J.M.; Petris, M.J.; Erb, L.; Weisman, G.A. P2 × 7 receptor activation induces inflammatory responses in salivary gland epithelium. Am. J. Physiol. Cell. Physiol. 2012, 303, C790–C801. [Google Scholar] [CrossRef] [Green Version]
- Khalafalla, M.G.; Woods, L.T.; Camden, J.M.; Khan, A.A.; Limesand, K.H.; Petris, M.J.; Erb, L.; Weisman, G.A. P2 × 7 receptor antagonism prevents IL-1beta release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. J. Biol. Chem. 2017, 292, 16626–16637. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Ren, Y.; Reinach, P.S.; She, Y.; Xiao, B.; Hua, S.; Qu, J.; Chen, W. Reactive oxygen species activated NLRP3 inflammasomes prime environment-induced murine dry eye. Exp. Eye Res. 2014, 125, 1–8. [Google Scholar] [CrossRef]
- Seror, R.; Ravaud, P.; Bowman, S.J.; Baron, G.; Tzioufas, A.; Theander, E.; Gottenberg, J.E.; Bootsma, H.; Mariette, X.; Vitali, C.; et al. EULAR Sjogren’s syndrome disease activity index: Development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1103–1109. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y.; Lee, G.H. Enhanced expression of NLRP3 inflammasome-related inflammation in peripheral blood mononuclear cells in Sjogren’s syndrome. Clin. Chim. Acta 2017, 474, 147–154. [Google Scholar] [CrossRef]
- Baldini, C.; Santini, E.; Rossi, C.; Donati, V.; Solini, A. The P2 × 7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin’s lymphoma in Sjogren’s syndrome: A prospective, observational, single-centre study. J. Intern. Med. 2017, 282, 175–186. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Arbeit, R.; Jiang, W.; Ortenzio, F.S.; Sullivan, T.; Krueger, J.G. Suppression of molecular inflammatory pathways by toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE 2013, 8, e84634. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Zhang, F.; Yin, H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol. Ther. 2013, 138, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balak, D.M.; van Doorn, M.B.; Arbeit, R.D.; Rijneveld, R.; Klaassen, E.; Sullivan, T.; Brevard, J.; Thio, H.B.; Prens, E.P.; Burggraaf, J.; et al. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin. Immunol. 2017, 174, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Barrera, M.J.; Aguilera, S.; Castro, I.; Matus, S.; Carvajal, P.; Molina, C.; González, S.; Jara, D.; Hermoso, M.; González, M.J. Tofacitinib counteracts IL-6 overexpression induced by deficient autophagy: Implications in Sjogren’s syndrome. Rheumatology 2020. [Google Scholar] [CrossRef]
- You, H.; Xu, D.; Zhao, J.; Li, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. JAK Inhibitors: Prospects in Connective Tissue Diseases. Clin. Rev. Allergy Immunol. 2020, 59, 334–351. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Ravaud, P.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Goeb, V.; Larroche, C.; Dubost, J.J.; Rist, S.; Saraux, A.; et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjögren syndrome: The JOQUER randomized clinical trial. JAMA 2014, 312, 249–258. [Google Scholar] [CrossRef]
- Yoon, C.H.; Lee, H.J.; Lee, E.Y.; Lee, E.B.; Lee, W.W.; Kim, M.K.; Wee, W.R. Effect of hydroxychloroquine treatment on dry eyes in subjects with primary Sjögren’s syndrome: A double-blind randomized control study. J. Korean Med. Sci. 2016, 31, 1127–1135. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimizu, T.; Nakamura, H.; Kawakami, A. Role of the Innate Immunity Signaling Pathway in the Pathogenesis of Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 3090. https://doi.org/10.3390/ijms22063090
Shimizu T, Nakamura H, Kawakami A. Role of the Innate Immunity Signaling Pathway in the Pathogenesis of Sjögren’s Syndrome. International Journal of Molecular Sciences. 2021; 22(6):3090. https://doi.org/10.3390/ijms22063090
Chicago/Turabian StyleShimizu, Toshimasa, Hideki Nakamura, and Atsushi Kawakami. 2021. "Role of the Innate Immunity Signaling Pathway in the Pathogenesis of Sjögren’s Syndrome" International Journal of Molecular Sciences 22, no. 6: 3090. https://doi.org/10.3390/ijms22063090
APA StyleShimizu, T., Nakamura, H., & Kawakami, A. (2021). Role of the Innate Immunity Signaling Pathway in the Pathogenesis of Sjögren’s Syndrome. International Journal of Molecular Sciences, 22(6), 3090. https://doi.org/10.3390/ijms22063090