
SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome




Abstract
:1. Introduction
1.1. Fibrosis in Sjögren’s Syndrome
1.2. Epithelial-Mesenchymal Transition (EMT) and Fibrosis
1.3. TGF-β, Master Regulator of EMT-Dependent Fibrosis
2. TGF-β Signal Transduction
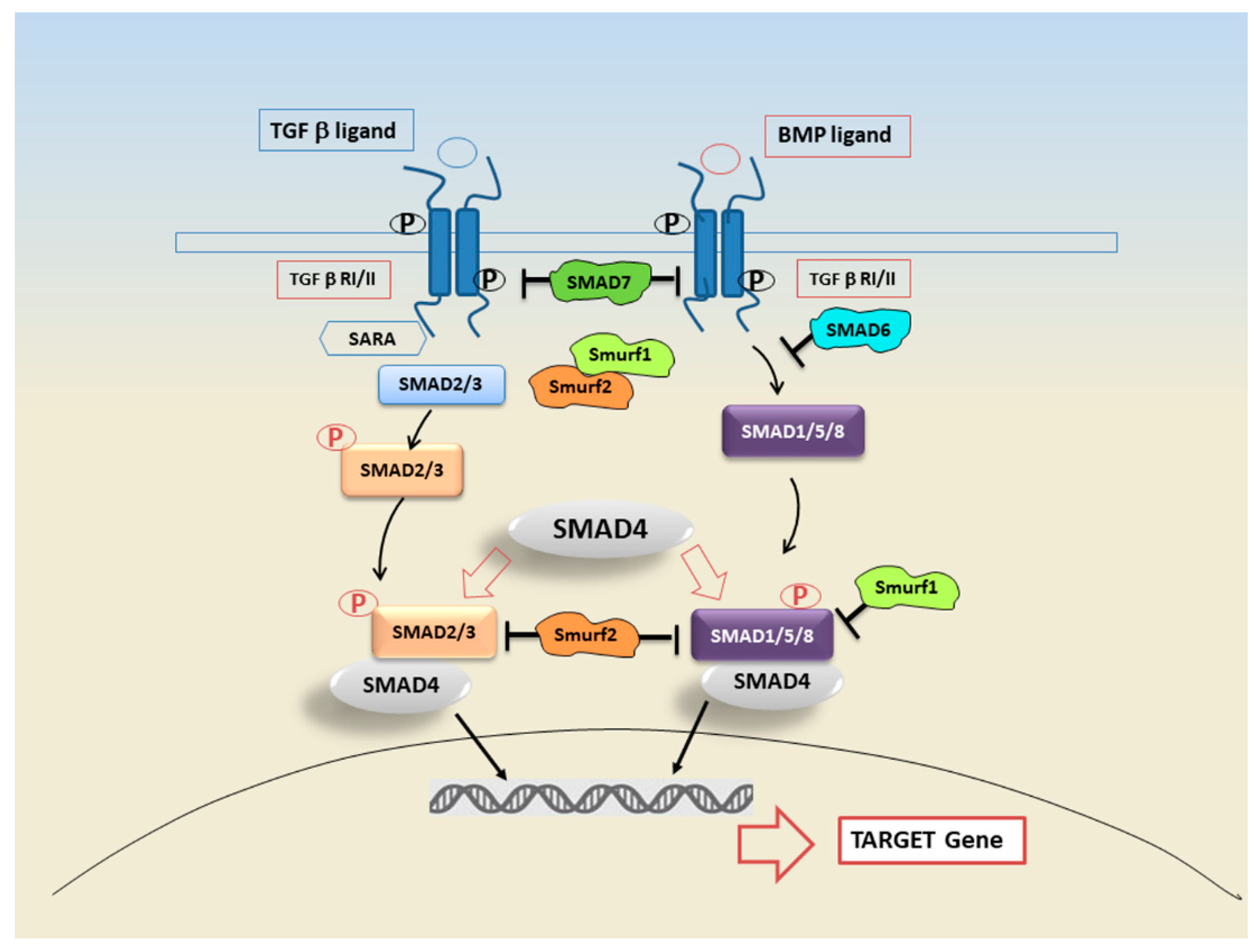
3. SMAD Proteins and Their Role in Signal Transduction
4. SMAD and Non-SMAD Pathways in TGF-β Signalling
5. SMADs Signalling Pathways Activated by TGF-β1-Dependent EMT in pSS
6. Inflammatory Mediators Trigger EMT in pSS through the Activation of TGF-β1/SMADs Canonical and Non-Canonical Pathways
7. Future Perspectives on Alternative Molecular Mechanisms Mediated by SMAD in SS
7.1. MicroRNAs and TGF-β/SMAD Signalling in SS
7.2. SMAD7 Functions as Inhibitor of JAK/STAT Pathway in SS
7.3. Potential Correlation of BMP6 with SMADs Phosphorylation in SS
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. J. Clin. Med. 2020, 9, 2299. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, S.; Adams, J.; de Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2021, 5, 1–26. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardet, A.; Zheng, T.S.; Viney, J.L. Genetic architecture of human fibrotic diseases: Disease risk and disease progression. Front Pharmacol. 2013, 4, 159. [Google Scholar] [CrossRef]
- Hsieh, C.; Chang, A.; Brandt, D.; Guttikonda, R.; Utset, T.O.; Clark, M.R. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res. 2011, 6, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Skopouli, F.N.; Li, L.; Boumba, D.; Stefanaki, S.; Hanel, K.; Moutsopoulos, H.M.; Krilis, S.A. Association of mast cells with fibrosis and fatty infiltration in the minor salivary glands of patients with Sjögren’s syndrome. Clin. Exp. Rheumatol. 1998, 1, 63–65. [Google Scholar]
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 112, 80–88. [Google Scholar]
- McArthur, C.P.; Daniels, P.J.; Kragel, P.; Howard, P.F.; Julian, L. Sjogren’s syndrome salivary gland immunopathology: Increased laminin expression precedes lymphocytic infiltration. J. Autoimmun. 1997, 10, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; de Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W. Epithelial-mesenchymal transition in human cancer: Comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol. Ther. 2015, 150, 33–46. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inan, S.; Hayran, M. Cell Signaling Pathways Related to Epithelial Mesenchymal Transition in Cancer Metastasis. Crit. Rev. Oncog. 2019, 24, 47–54. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Lorusso, L.; Tamma, R.; Ingravallo, G.; Ribatti, D.; Lisi, S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren’s syndrome. Clin. Exp. Immunol. 2019, 198, 261–272. [Google Scholar] [CrossRef]
- Sisto, M.; Tamma, R.; Ribatti, D.; Lisi, S. IL-6 Contributes to the TGF-β1-Mediated Epithelial to Mesenchymal Transition in Human Salivary Gland Epithelial Cells. Arch. Immunol. Ther. Exp. 2020, 68, 27. [Google Scholar] [CrossRef]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef]
- Scimone, C.; Bramanti, P.; Ruggeri, A.; Donato, L.; Alafaci, C.; Crisafulli, C.; Mucciardi, M.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. CCM3/SERPINI1 bidirectional promoter variants in patients with cerebral cavernous malformations: A molecular and functional study. BMC Med. Genet. 2016, 17, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, C.; Bramanti, P.; Scimone, C.; Donato, L.; Alafaci, C.; D’Angelo, R.; Sidoti, A. Relevance of CCM gene polymorphisms for clinical management of sporadic cerebral cavernous malformations. J. Neurol. Sci. 2017, 380, 31–37. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Katsarou, Z.; Bostantjopoulou, S.; D’Angelo, R.; Sidoti, A. Two Novel KRIT1 and CCM2 Mutations in Patients Affected by Cerebral Cavernous Malformations: New Information on CCM2 Penetrance. Front. Neurol. 2018, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Donato, L.; Alafaci, C.; Granata, F.; Rinaldi, C.; Longo, M.; D’Angelo, R.; Sidoti, A. High-Throughput Sequencing to Detect Novel Likely Gene-Disrupting Variants in Pathogenesis of Sporadic Brain Arteriovenous Malformations. Front. Genet. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Granata, F.; Longo, M.; Mormina, E.; Turiaco, C.; Caragliano, A.A.; Donato, L.; Sidoti, A.; D’Angelo, R. Germline Mutation Enrichment in Pathways Controlling Endothelial Cell Homeostasis in Patients with Brain Arteriovenous Malformation: Implication for Molecular Diagnosis. Int. J. Mol. Sci. 2020, 21, 4321. [Google Scholar] [CrossRef]
- Speca, S.; Giusti, I.; Rieder, F.; Latella, G. Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 2012, 18, 3635–3661. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; Ribatti, D. The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands. Histochem. Cell Biol. 2018, 150, 133–147. [Google Scholar] [CrossRef]
- Roberts, A.B.; Russo, A.; Felici, A.; Flanders, K.C. SMAD3: A key player in pathogenetic mechanisms dependent on TGF-beta. Ann. N. Y. Acad. Sci. 2003, 995, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Massagué, J. The transforming growth factor-β family. Ann. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-β superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 10, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [Green Version]
- Lucas, P.J.; Kim, S.-J.; Mackall, C.L.; Telford, W.G.; Chu, Y.-W.; Hakim, F.T.; Gress, R.E. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-β dominant-negative receptor transgenic mice. Blood 2006, 108, 2789–2795. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, T.; Olofsson, A.; Moren, A.; Wernstedt, C.; Hellman, U.; Miyazono, K.; Claesson-Welsh, L.; Heldin, C.H. TGF-beta 1 binding protein: A component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell 1990, 61, 1051–1061. [Google Scholar] [CrossRef]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF β activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massague, J. The TGF β receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-β/SMAD signaling. FEBS Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef] [Green Version]
- Gu, A.D.; Wang, Y.; Lin, l.; Zhang, S.S.; Yisong, Y. Wan Requirements of transcription factor SMAD-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc. Natl. Acad. Sci. USA 2012, 109, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, X.; Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 2014, 5, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. SMAD-dependent and SMAD-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-SMAD pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Sekelsky, J.J.; Newfeld, S.; Raftery, L.A.; Chartoff, E.H.; Gelbart, W.M. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 1995, 139, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Gelbart, W. The decapentaplegic gene: A TGF-β homolog controlling pattern formation in Drosophila. Development 1989, 107, 65–74. [Google Scholar]
- Ferguson, E.; Anderson, K. Decapentaplegic acts as a morphogen to organize dorsal-ventral patern in the Drosophila embryo. Cell 1992, 54, 95–104. [Google Scholar]
- Derynck, R.; Gelbart, W.M.; Harland, R.M.; Heldin, C.H.; Kern, S.E.; Massagué, J.; Melton, D.A.; Mlodzik, M.; Padgett, R.W.; Roberts, A.B.; et al. Nomenclature: Vertebrate mediators of TGF-beta family signals. Cell 1996, 87, 173. [Google Scholar] [CrossRef] [Green Version]
- Patterson, G.I.; Padgett, R.W. TGF β-related pathways: Roles in Caenorhabditis elegans development. Trends Genet. 2000, 16, 27–33. [Google Scholar] [CrossRef]
- Whitman, M. SMADs and early developmental signaling by the TGF-β superfamily. Genes Dev. 1998, 12, 2445–2462. [Google Scholar] [CrossRef] [Green Version]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. The regulation of TGF-β signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Dijke, P.; Heldin, C.H. The SMAD Family. In SMAD Signal Transduction; Ten Dijke, P., Heldin, C.H., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 5, pp. 1–13. [Google Scholar]
- Attisano, L.; Lee-Hoeflich, S.T. The SMADs. Genome Biol. 2001, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C.; Kim, E.S.; Roberts, A.B. Immunohistochemical expression of SMADs1–6 in the 15-day gestation mouse embryo: Signaling by BMPs and TGF-βs. Dev. Dyn. 2001, 220, 141–154. [Google Scholar] [CrossRef]
- Luukko, K.; Ylikorkala, A.; Mäkelä, T.P. Developmentally regulated expression of SMAD3, SMAD4, SMAD6 and SMAD7 involved in TGF-beta signaling. Mech. Dev. 2001, 101, 209–212. [Google Scholar] [CrossRef]
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of SMAD2 on Ser465 and Ser467 is required for SMAD2–SMAD4 complex formation and signaling. J. Biol. Chem. 1997, 272, 7678–7685. [Google Scholar] [CrossRef] [Green Version]
- Souchelnytskyi, S.; Tamaki, K.; Engstrom, U.; Wernstedt, C.; Ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of SMAD2 mediates interaction with SMAD4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef] [Green Version]
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massagué, J. Partnership between DPC4 and SMAD proteins in TGF-beta signalling pathways. Nature 1996, 383, 832–836. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGF-β superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Soond, S.M.; Chantry, A. How ubiquitination regulates the TGF-β signalling pathway: New insights and new players: New isoforms of ubiquitin-activating enzymes in the E1-E3 families join the game. Bioessays 2011, 33, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Soond, S.M.; Chantry, A. Selective targeting of activating and inhibitory SMADs by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGF-β signalling and EMT. Oncogene 2011, 30, 2451–2462. [Google Scholar] [CrossRef] [Green Version]
- Wahl, L.C.; Watt, J.E.; Yim, H.T.T.; de Bourcier, D.; Tolchard, J.; Soond, S.M.; Blumenschein, T.M.A.; Chantry, A. SMAD7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms. Int. J. Mol. Sci. 2019, 20, 4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Ribatti, D.; Lisi, S. TGF-β1-SMAD canonical and -Erk non-canonical pathways participate in interleukin-17-induced epithelial—Mesenchymal transition in Sjögren’s syndrome. Lab. Investig. 2020, 100, 824–836. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Kalluri, R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front. Biosci. 2008, 13, 6991–6998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prud’homme, G. Pathobiology of transforming growth factor β in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, 5183. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.; Lin, X.; Feng, X. Phospho-control of TGF-β superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandl, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. IKKα controls canonical TGF-β—SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in Panc1 cells. J. Cell Sci. 2010, 123, 4231–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/SMAD3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Yang, X.; Glick, A.B.; Weinstein, M.; Letterio, J.L.; Mizel, D.E.; Anzano, M.; Greenwell-Wild, T.; Wahl, S.M.; Deng, C.; et al. Mice lacking SMAD3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999, 1, 260–266. [Google Scholar] [CrossRef]
- Hoot, K.E.; Lighthall, J.; Han, G.; Lu, S.L.; Li, A.; Ju, W.; Kulesz-Martin, M.; Bottinger, E.; Wang, X.J. Keratinocyte-specific SMAD2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J. Clin. Investig. 2008, 118, 2722–2732. [Google Scholar] [CrossRef] [Green Version]
- Ju, W.; Ogawa, A.; Heyer, J.; Nierhof, D.; Yu, L.; Kucherlapati, R.; Shafritz, D.A.; Böttinger, E.P. Deletion of SMAD2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol. Cell. Biol. 2006, 26, 654–667. [Google Scholar] [CrossRef] [Green Version]
- Takano, S.; Kanai, F.; Jazag, A.; Ijichi, H.; Yao, J.; Ogawa, H.; Enomoto, N.; Omata, M.; Nakao, A. SMAD4 is essential for down-regulation of E-cadherin induced by TGF-β in pancreatic cancer cell line PANC-1. J. Biochem. 2007, 141, 345–351. [Google Scholar] [CrossRef]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Löwik, C.; van der Pluijm, G.; Ten Dijke, P. The tumor suppressor SMAD4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef] [Green Version]
- Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; DePinho, R.A. SMAD4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein SMAD7 associates with the TGF beta receptor and functions as an antagonist of TGF beta signalling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Afrakht, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of SMAD7, a TGF-β-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory SMADs. Cold Spring Harb. Perspect. Biol. 2016, 9, a022095. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through SMADs. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.P.; Li, Q.Q.; Cao, X.X.; Chen, Q.; Zhao, Z.H.; Diao, Z.Q.; Xu, Z.D. The effect of TGF-β1 and SMAD7 gene transfer on the phenotypic changes of rat alveolar epithelial cells. Cell Mol. Biol. Lett. 2007, 12, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific SMAD7 expression attenuates TGF-β-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659. [Google Scholar] [CrossRef] [Green Version]
- Boirivant, M.; Pallone, F.; Di Giacinto, C.; Fina, D.; Monteleone, I.; Marinaro, M.; Caruso, R.; Colantoni, A.; Palmieri, G.; Sanchez, M.; et al. Inhibition of SMAD7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 2006, 131, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, S.; Bevivino, G.; Monteleone, G. Mongersen, an oral SMAD7 antisense oligonucleotide, in patients with active Crohn’s disease. Ther. Adv. Gastroenterol. 2016, 9, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.E. Non-SMAD Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGF-β cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yun, J.S.; Han, D.; Yook, J.I.; Kim, H.S.; Cho, E.S. TGF-β Pathway in Salivary Gland Fibrosis. Int. J. Mol. Sci. 2020, 21, 9138. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Tamma, R.; Ribatti, D.; Lisi, S. The TGF-β1 Signaling Pathway as an Attractive Target in the Fibrosis Pathogenesis of Sjögren’s Syndrome. Mediat. Inflamm. 2018, 2018, 1965935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Mitolo, V.; D’Amore, M. Expression of pro-inflammatory TACE-TNF-α-amphiregulin axis in Sjögren’s syndrome salivary glands. Histochem. Cell Biol. 2010, 134, 345–353. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab. Investig. 2012, 92, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 71, 295–301. [Google Scholar] [CrossRef]
- Ciccia, F.; Giardina, A.; Rizzo, A.; Guggino, G.; Cipriani, P.; Carubbi, F.; Giacomelli, R.; Triolo, G. Rituximab modulates the expression of IL-22 in the salivary glands of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2012, 72, 782–783. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- de Morales, J.M.G.R.; Puig, L.; Daudén, E.; Cañete, J.D.; Pablos, J.L.; Martín, A.O.; Juanatey, C.G.; Adán, A.; Montalbán, X.; Borruel, N.; et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef]
- Li, R.; Chung, A.C.; Yu, X.; Lan, H.Y. MicroRNAs in diabetic kidney disease. Int. J. Endocrinol. 2014, 2014, 593956. [Google Scholar] [CrossRef] [Green Version]
- Vettori, S.; Gay, S.; Distler, O. Role of microRNAs in fibrosis. Open Rheumatol. J. 2012, 6, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chung, A.C.; Dong, Y.; Yang, W.; Zhong, X.; Lan, H.Y. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-β/SMAD3-Azin1 pathway. Kidney Int. 2013, 84, 1129–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, Z.; Olthof, E.D.; Drexhage, H.A.; Versnel, M.A. Monocyte gene expression signatures in rheumatic diseases: Biomarkers for disease activity and tools for diagnosis and classification. Open Arthritis J. 2010, 3, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Alevizos, I.; Alexander, S.; Turner, R.J.; Illei, G.G. MicroRNA expression profiles as biomarkers of minor salivary gland inflammation and dysfunction in Sjogren’s syndrome. Arthritis Rheum. 2011, 63, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapsogeorgou, E.K.; Gourzi, V.C.; Manoussakis, M.N.; Moutsopoulos, H.M.; Tzioufas, A.G. Cellular microRNAs (miRNAs) and Sjogren’s syndrome: Candidate regulators of autoimmune response and autoantigen expression. J. Autoimmun. 2011, 37, 129–135. [Google Scholar] [CrossRef]
- Reale, M.; D’Angelo, C.; Costantini, E.; Laus, M.; Moretti, A.; Croce, A. MicroRNA in Sjögren’s Syndrome: Their Potential Roles in Pathogenesis and Diagnosis. J. Immunol. Res. 2018, 2018, 7510174. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.E.; Choi, K.; Chan, A.L.; Lee, Y.J.; Reeves, W.H.; Bubb, M.R.; Stewart, C.M.; Cha, S. Sjogren’s syndrome-associated microRNAs in CD14(+) monocytes unveils targeted TGF beta signaling. Arthritis Res. Ther. 2016, 18, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-Q.; Papp, G.; Póliska, S.; Szabó, K.; Tarr, T.; Bálint, B.L.; Szodoray, P.; Zeher, M. MicroRNA expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary Sjögren’s syndrome. PLoS ONE 2017, 12, e0174585. [Google Scholar] [CrossRef]
- Charras, A.; Arvaniti, P.; Le Dantec, C.; Dalekos, G.N.; Zachou, K.; Bordron, A.; Renaudineau, Y. JAK Inhibitors and Oxidative Stress Control. Front. Immunol. 2019, 10, 2814. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Tilakaratne, W.M.; Seoudi, N.; Bombardieri, M.; Bergmeier, L.; Tappuni, A.R.; Fortune, F. Dysregulation of the suppressor of cytokine signalling 3-signal transducer and activator of transcription-3 pathway in the aetiopathogenesis of Sjögren’s syndrome. Clin. Exp. Immunol. 2014, 177, 618–629. [Google Scholar] [CrossRef]
- Pringle, S.; Wang, X.; Verstappen, G.M.P.J.; Terpstra, J.H.; Zhang, C.K.; He, A.; Patel, V.; Jones, R.E.; Baird, D.M.; Spijkervet, F.K.L.; et al. Salivary Gland Stem Cells Age Prematurely in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2018, 71, 133–142. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hill, C.S. Tgf-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGF-β signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Kahata, K.; Maturi, V.; Moustakas, A. TGF-β Family Signaling in Ductal Differentiation and Branching Morphogenesis. Cold Spring Harb. Perspect. Biol. 2017, 10, a031997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-βsignaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Yu, Y.; Gu, S.; Li, W.; Sun, C.; Chen, F.; Xiao, M.; Wang, L.; Xu, D.; Li, Y.; Ding, C.; et al. Smad7 enables STAT3 activation and promotes pluripotency independent of TGF-β signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 10113–10118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, K.; Inagaki, Y.; Fujimori, K.; Nakao, A.; Kaneko, H.; Nakatsuka, I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with SMAD3. J. Biol. Chem. 2003, 278, 43470–43479. [Google Scholar] [CrossRef] [Green Version]
- Horvath, C.M. The Jak-STAT pathway stimulated by interferon γ. Sci. STKE 2004, 2004, tr8. [Google Scholar] [CrossRef]
- Ulloa, L.; Doody, J.; Massagué, J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature 1999, 397, 710–713. [Google Scholar] [CrossRef]
- de Ceuninck van Capelle, C.; Spit, M.; Ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Crit Rev Biochem Mol Biol. 2020, 55, 691–715. [Google Scholar] [CrossRef]
- Yin, H.; Kalra, L.; Lai, Z.; Guimaro, M.C.; Aber, L.; Warner, B.M.; Michael, D.; Zhang, N.; Cabrera-Perez, J.; Karim, A.; et al. Inhibition of bone morphogenetic protein 6 receptors ameliorates Sjögren’s syndrome in mice. Sci. Rep. 2020, 10, 2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Cabrera-Perez, J.; Lai, Z.; Michael, D.; Weller, M.; Swaim, W.D.; Liu, X.; Catalán, M.A.; Rocha, E.M.; Ismail, N.; et al. Association of bone morphogenetic protein 6 with exocrine gland dysfunction in patients with Sjögren’s syndrome and in mice. Arthritis Rheum. 2013, 65, 3228–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Su, Y.; Hu, L.; Cain, A.; Gu, Y.; Liu, B.; Wu, R.; Wang, S.; Wang, H. Effect of Bone Morphogenetic Protein 6 on Immunomodulatory Functions of Salivary Gland-Derived Mesenchymal Stem Cells in Sjögren’s Syndrome. Stem Cells Dev. 2018, 27, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Yin, H.; Cabrera-Pérez, J.; Guimaro, M.C.; Afione, S.; Michael, D.G.; Glenton, P.; Patel, A.; Swaim, W.D.; Zheng, C.; et al. Aquaporin gene therapy corrects Sjögren’s syndrome phenotype in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5694–5699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinfeld, S.; Cogan, E.; King, L.S.; Agre, P.; Kiss, R.; Delporte, C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjögren’s syndrome patients. Lab. Investig. 2001, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy [93] | Potential Therapy |
|---|---|
| Block activation of TGF-β receptors inhibitors | Small molecule inhibitors |
| Block coactivator recruitment and function | Aptamers (Trx-SARA) |
| Block ligand production or activity | Isotype-specific neutralizing antibodies |
| Soluble TβR1-3 receptors | |
| Antibodies to avß6 integrin | |
| Natural TGF-β binding proteins (eg. Decorin) | |
| Nucleic acid-based (antisense, ribozyme, siRNA) | |
| Inhibition of the SMAD Pathway | Physiologic endogenous inhibitor Smad7 |
| halofuginone (HT-100) | |
| SIS3, targets SMAD3 phosphorylation | |
| Adenovirus vector gene transfer of SMAD7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisto, M.; Ribatti, D.; Lisi, S. SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 3203. https://doi.org/10.3390/ijms22063203
Sisto M, Ribatti D, Lisi S. SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome. International Journal of Molecular Sciences. 2021; 22(6):3203. https://doi.org/10.3390/ijms22063203
Chicago/Turabian StyleSisto, Margherita, Domenico Ribatti, and Sabrina Lisi. 2021. "SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome" International Journal of Molecular Sciences 22, no. 6: 3203. https://doi.org/10.3390/ijms22063203
APA StyleSisto, M., Ribatti, D., & Lisi, S. (2021). SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome. International Journal of Molecular Sciences, 22(6), 3203. https://doi.org/10.3390/ijms22063203