
Sexual Dimorphism in Glucocorticoid Stress Response


{kind=link}
{kind=link}
Abstract
:1. Introduction
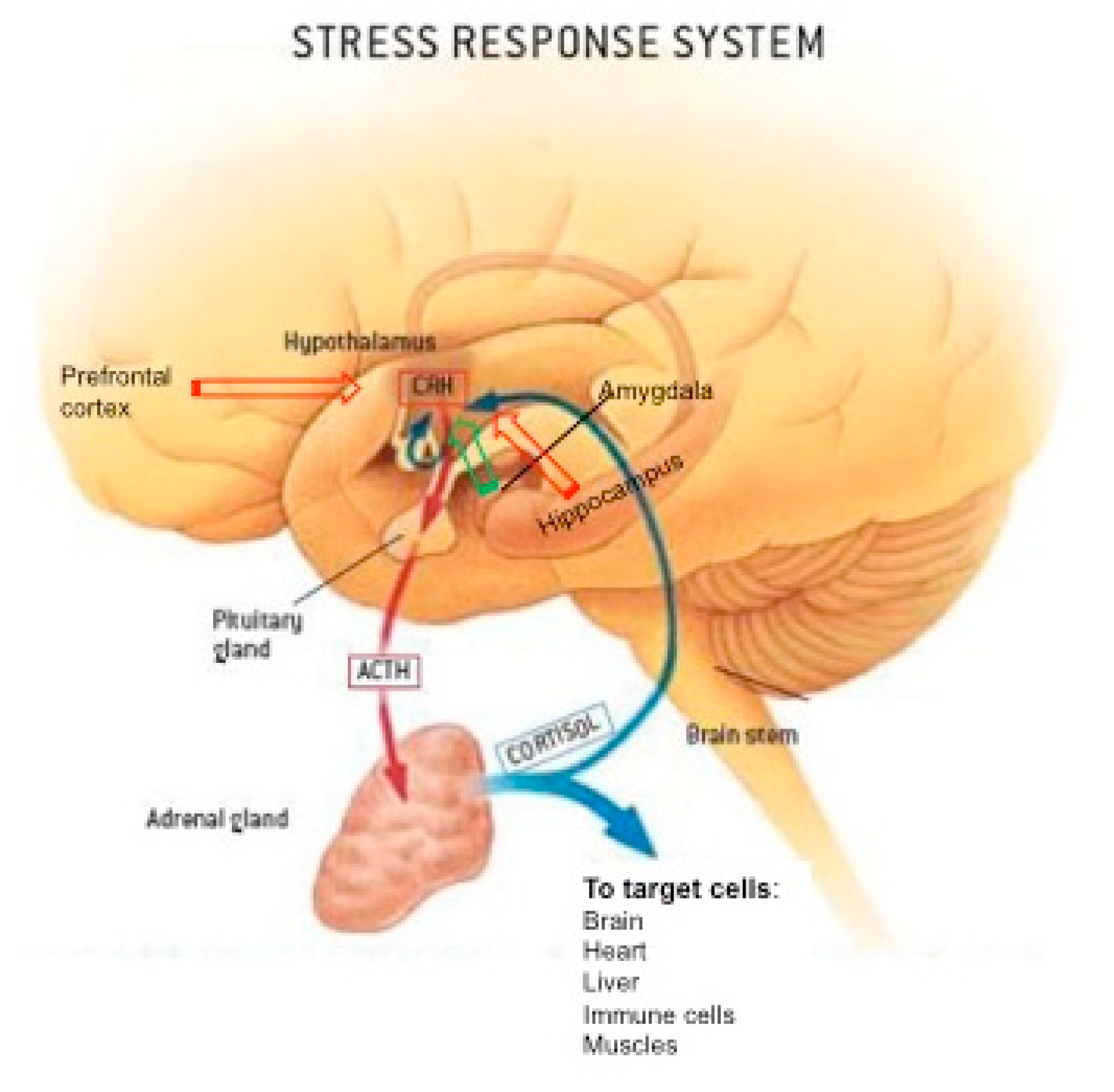
2. Stress Response System, the Hypothalamic-Pituitary-Adrenal Axis, and GC Hormones
3. Genetic Causes of Sexual Dimorphism
4. Sex Dimorphism Related to Gonadal Hormones across Development
4.1. Neonatal Period
4.2. Puberty Period
4.3. Adulthood
4.4. Molecular Aspects
5. Sex-Specific Programming Effects of Stress on the HPA Axis
5.1. Perinatal Environment
5.2. Adolescent Period
6. Discussion
Funding
Conflicts of Interest
References
- McEwen, B.S.; Akil, H. Revisiting the Stress Concept: Implications for Affective Disorders. J. Neurosci. 2020, 40, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef]
- Moisan, M.P. CBG: A cortisol reservoir rather than a transporter. Nat. Rev. Endocrinol. 2013, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joels, M.; Karst, H.; DeRijk, R.; De Kloet, E.R. The coming out of the brain mineralocorticoid receptor. Trends. Neurosci. 2008, 31, 1–7. [Google Scholar] [CrossRef]
- Jiang, C.-L.; Liu, L.; Tasker, J.G. Why do we need nongenomic glucocorticoid mechanisms? Front. Neuroendocrinol. 2014, 35, 72–75. [Google Scholar] [CrossRef]
- Strehl, C.; Buttgereit, F. Unraveling the functions of the membrane-bound glucocorticoid receptors: First clues on origin and functional activity. Ann. N. Y. Acad. Sci. 2014, 1318, 1–6. [Google Scholar] [CrossRef]
- Malkoski, S.P.; Dorin, R.I. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol. Endocrinol. 1999, 13, 1629–1644. [Google Scholar] [CrossRef]
- Guardiola-Diaz, H.M.; Kolinske, J.S.; Gates, L.H.; Seasholtz, A.F. Negative glucorticoid regulation of cyclic adenosine 3′, 5′-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol. Endocrinol. 1996, 10, 317–329. [Google Scholar]
- Drouin, J. 60 YEARS OF POMC: Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef]
- Joels, M.; Baram, T.Z. The neuro-symphony of stress. Nat. Rev. Neurosci. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [Green Version]
- Simmler, M.C.; Rouyer, F.; Vergnaud, G.; Nyström-Lahti, M.; Ngo, K.Y.; de la Chapelle, A.; Weissenbach, J. Pseudoautosomal DNA sequences in the pairing region of the human sex chromosomes. Nature 1985, 317, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Gendrel, A.-V.; Heard, E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu. Rev. Cell Dev. Biol. 2014, 30, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R. Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend. Med. 2007, 4, 97–105. [Google Scholar] [CrossRef]
- Raznahan, A.; Disteche, C.M. X-chromosome regulation and sex differences in brain anatomy. Neurosci. Biobehav. Rev. 2021, 120, 28–47. [Google Scholar] [CrossRef]
- Hessl, D.; Glaser, B.; Dyer-Friedman, J.; Blasey, C.; Hastie, T.; Gunnar, M.; Reiss, A.L. Cortisol and behavior in fragile X syndrome. Psychoneuroendocrinology 2002, 27, 855–872. [Google Scholar] [CrossRef]
- Gregg, C.; Zhang, J.; Butler, J.E.; Haig, D.; Dulac, C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 682–685. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.M.; Nugent, B.M.; Lenz, K.M. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nat. Rev. Neurosci. 2017, 18, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.A.; Jordan, C.L.; Breedlove, S.M. Sexual differentiation of the vertebrate nervous system. Nat. Neurosci. 2004, 7, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Minni, A.M.; de Medeiros, G.F.; Helbling, J.C.; Duittoz, A.; Marissal-Arvy, N.; Foury, A.; De Smedt-Peyrusse, V.; Pallet, V.; Moisan, M.P. Role of corticosteroid binding globulin in emotional reactivity sex differences in mice. Psychoneuroendocrinology 2014, 50, 252–263. [Google Scholar] [CrossRef]
- Toews, J.N.C.; Hammond, G.L.; Viau, V. Liver at the nexus of rat postnatal HPA axis maturation and sexual dimorphism. J. Endocrinol. 2021, 248, R1–R17. [Google Scholar] [CrossRef]
- Chowen, J.A.; Frago, L.M.; Argente, J. The regulation of GH secretion by sex steroids. Eur. J. Endocrinol. 2004, 151 (Suppl. 3), U95–U100. [Google Scholar] [CrossRef] [Green Version]
- Jansson, J.O.; Oscarsson, J.; Mode, A.; Ritzén, E.M. Plasma growth hormone pattern and androgens influence the levels of corticosteroid-binding globulin in rat serum. J. Endocrinol. 1989, 122, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Zuloaga, D.G.; Heck, A.L.; De Guzman, R.M.; Handa, R.J. Roles for androgens in mediating the sex differences of neuroendocrine and behavioral stress responses. Biol. Sex Differ. 2020, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.M.; Furey, B.F.; Child, M.; Sawyer, M.J.; Donohue, S.M. Neonatal sex hormones have ‘organizational’ effects on the hypothalamic-pituitary-adrenal axis of male rats. Dev. Brain Res. 1998, 105, 295–307. [Google Scholar] [CrossRef]
- Seale, J.V.; Wood, S.A.; Atkinson, H.C.; Lightman, S.L.; Harbuz, M.S. Organizational role for testosterone and estrogen on adult hypothalamic-pituitary-adrenal axis activity in the male rat. Endocrinology 2005, 146, 1973–1982. [Google Scholar] [CrossRef]
- Bingham, B.; Wang, N.X.R.; Innala, L.; Viau, V. Postnatal aromatase blockade increases c-fos mRNA responses to acute restraint stress in adult male rats. Endocrinology 2012, 153, 1603–1608. [Google Scholar] [CrossRef] [Green Version]
- Motta-Mena, N.V.; Puts, D.A. Endocrinology of human female sexuality, mating, and reproductive behavior. Horm. Behav. 2017, 91, 19–35. [Google Scholar] [CrossRef]
- Jaffe, C.A.; Ocampo-Lim, B.; Guo, W.; Krueger, K.; Sugahara, I.; DeMott-Friberg, R.; Bermann, M.; Barkan, A.L. Regulatory mechanisms of growth hormone secretion are sexually dimorphic. J. Clin. Investig. 1998, 102, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Tschöp, M.; Lahner, H.; Feldmeier, H.; Grasberger, H.; Morrison, K.M.; Janssen, O.E.; Attanasio, A.F.; Strasburger, C.J. Effects of growth hormone replacement therapy on levels of cortisol and cortisol-binding globulin in hypopituitary adults. Eur. J. Endocrinol. 2000, 143, 769–773. [Google Scholar] [CrossRef]
- Romeo, R.D. The metamorphosis of adolescent hormonal stress reactivity: A focus on animal models. Front. Neuroendocrinol. 2018, 49, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Evuarherhe, O.; Leggett, J.D.; Waite, E.J.; Kershaw, Y.M.; Atkinson, H.C.; Lightman, S.L. Organizational role for pubertal androgens on adult hypothalamic-pituitary-adrenal sensitivity to testosterone in the male rat. J. Physiol. 2009, 587, 2977–2985. [Google Scholar] [CrossRef] [PubMed]
- Evuarherhe, O.; Leggett, J.; Waite, E.; Kershaw, Y.; Lightman, S. Reversal of the hypothalamo-pituitary-adrenal response to oestrogens around puberty. J. Endocrinol. 2009, 202, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Hii, H.L.; Pennell, C.E.; McKeague, I.W.; de Kloet, E.R.; Lye, S.; Stanley, F.J.; Mattes, E.; Foster, J.K. Analysis of baseline hypothalamic-pituitary-adrenal activity in late adolescence reveals gender specific sensitivity of the stress axis. Psychoneuroendocrinology 2013, 38, 1271–1280. [Google Scholar] [CrossRef]
- Singer, N.; Sommer, M.; Wüst, S.; Kudielka, B.M. Effects of gender and personality on everyday moral decision-making after acute stress exposure. Psychoneuroendocrinology 2021, 124, 105084. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.W.; Ein, N.; Peck, K.; Huang, V.; Pruessner, J.C.; Vickers, K. Sex differences in salivary cortisol reactivity to the Trier Social Stress Test (TSST): A meta-analysis. Psychoneuroendocrinology 2017, 82, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.J.; Nenke, M.A.; Rankin, W.; Lewis, J.G.; Torpy, D.J. Corticosteroid-Binding Globulin: A Review of Basic and Clinical Advances. Horm. Metab. Res. 2016, 48, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki-Sekino, A.; Mano-Otagiri, A.; Ohata, H.; Yamauchi, N.; Shibasaki, T. Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological stress in rats. Psychoneuroendocrinology 2009, 34, 226–237. [Google Scholar]
- Turner, B.B. Sex difference in glucocorticoid binding in rat pituitary is estrogen dependent. Life Sci. 1990, 46, 1399–1406. [Google Scholar] [CrossRef]
- Solomon, M.B.; Loftspring, M.; de Kloet, A.D.; Ghosal, S.; Jankord, R.; Flak, J.N.; Wulsin, A.C.; Krause, E.G.; Zhang, R.; Rice, T.; et al. Neuroendocrine Function After Hypothalamic Depletion of Glucocorticoid Receptors in Male and Female Mice. Endocrinology 2015, 156, 2843–2853. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, D.; Padmanabhan, V.; Sargis, R.M. Stress, Sex, and Sugar: Glucocorticoids and Sex-Steroid Crosstalk in the Sex-Specific Misprogramming of Metabolism. J. Endocr. Soc. 2020, 4, bvaa087. [Google Scholar] [CrossRef]
- Dovey, J.L.; Vasudevan, N. Does GPER1 Play a Role in Sexual Dimorphism? Front. Endocrinol. (Lausanne) 2020, 11, 595895. [Google Scholar] [CrossRef]
- Bao, A.-M.; Fischer, D.F.; Wu, Y.-H.; Hol, E.M.; Balesar, R.; Unmehopa, U.A.; Zhou, J.-N.; Swaab, D.F. A direct androgenic involvement in the expression of human corticotropin-releasing hormone. Mol. Psychiatry 2006, 11, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, J.; Matsumoto, T.; Shiina, H.; Inoue, K.; Takada, I.; Ito, S.; Itoh, J.; Minematsu, T.; Sato, T.; Yanase, T. The pituitary function of androgen receptor constitutes a glucocorticoid production circuit. Mol. Cell Biol. 2007, 27, 4807–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, T.D.; Hinds, L.R.; Handa, R.J. The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha-androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J. Neurosci. 2006, 26, 1448–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, T.R.; Chung, W.C.J.; Hinds, L.R.; Handa, R.J. Estrogen receptor-beta mediates dihydrotestosterone-induced stimulation of the arginine vasopressin promoter in neuronal cells. Endocrinology 2007, 148, 3371–3382. [Google Scholar] [CrossRef]
- Hiroi, R.; Lacagnina, A.F.; Hinds, L.R.; Carbone, D.G.; Uht, R.M.; Handa, R.J. The androgen metabolite, 5α-androstane-3β,17β-diol (3β-diol), activates the oxytocin promoter through an estrogen receptor-β pathway. Endocrinology 2013, 154, 1802–1812. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.; Li, Q.-C.; Zhu, Q.-B.; Hu, S.-H.; Balesar, R.; Swaab, D.; Bao, A.M. Direct Involvement of Androgen Receptor in Oxytocin Gene Expression: Possible Relevance for Mood Disorders. Neuropsychopharmacology 2017, 42, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-N.; Zhu, H.; Meng, Q.-Y.; Zhou, J.-N. Estrogen receptor-alpha and -beta regulate the human corticotropin-releasing hormone gene through similar pathways. Brain. Res. 2008, 1223, 1–10. [Google Scholar] [CrossRef]
- Van de Stolpe, A.; Slycke, A.J.; Reinders, M.O.; Zomer, A.W.M.; Goodenough, S.; Behl, C.; Seasholtz, A.F.; van der Saag, P.T. Estrogen receptor (ER)-mediated transcriptional regulation of the human corticotropin-releasing hormone-binding protein promoter: Differential effects of ERalpha and ERbeta. Mol. Endocrinol. 2004, 18, 2908–2923. [Google Scholar] [CrossRef] [Green Version]
- Burrows, H.L.; Nakajima, M.; Lesh, J.S.; Goosens, K.A.; Samuelson, L.C.; Inui, A.; Camper, S.A.; Seasholtz, A.F. Excess corticotropin releasing hormone-binding protein in the hypothalamic-pituitary-adrenal axis in transgenic mice. J. Clin. Investig. 1998, 101, 1439–1447. [Google Scholar] [CrossRef]
- Jopek, K.; Celichowski, P.; Szyszka, M.; Tyczewska, M.; Milecka, P.; Malendowicz, L.K.; Rucinski, M. Transcriptome Profile of Rat Adrenal Evoked by Gonadectomy and Testosterone or Estradiol Replacement. Front. Endocrinol. (Lausanne) 2017, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Nader, N.; Raverot, G.; Emptoz-Bonneton, A.; Dechaud, H.; Bonnay, M.; Baudin, E.; Pugeat, M. Mitotane has an estrogenic effect on sex hormone-binding globulin and corticosteroid-binding globulin in humans. J. Clin. Endocrinol. Metab. 2006, 91, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- Shen, N.; Gong, J.; Wang, Y.; Tian, J.; Qian, J.; Zou, L.; Chen, W.; Zhu, B.; Lu, X.; Zhong, R. Integrative genomic analysis identifies that SERPINA6-rs1998056 regulated by FOXA/ERα is associated with female hepatocellular carcinoma. PLoS ONE 2014, 9, e107246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panton, K.K.; Mikkelsen, G.; Irgens, W.Ø.; Hovde, A.K.; Killingmo, M.W.; Øien, M.A.; Thorsby, P.M.; Asberg, A. New reference intervals for cortisol, cortisol binding globulin and free cortisol index in women using ethinyl estradiol. Scand. J. Clin. Lab. Investig. 2019, 79, 314–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patchev, V.K.; Shoaib, M.; Holsboer, F.; Almeida, O.F. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience 1994, 62, 265–271. [Google Scholar] [CrossRef]
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef]
- Mueller, B.R.; Bale, T.L. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol. Behav. 2007, 91, 55–65. [Google Scholar] [CrossRef]
- Darnaudéry, M.; Maccari, S. Epigenetic programming of the stress response in male and female rats by prenatal restraint stress. Brain. Res. Rev. 2008, 57, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Matthews, S.G. Prenatal stress modifies behavior and hypothalamic-pituitary-adrenal function in female guinea pig offspring: Effects of timing of prenatal stress and stage of reproductive cycle. Endocrinology 2008, 149, 6406–6415. [Google Scholar] [CrossRef] [Green Version]
- Thayer, Z.M.; Wilson, M.A.; Kim, A.W.; Jaeggi, A.V. Impact of prenatal stress on offspring glucocorticoid levels: A phylogenetic meta-analysis across 14 vertebrate species. Sci. Rep. 2018, 8, 4942. [Google Scholar] [CrossRef]
- Howerton, C.L.; Morgan, C.P.; Fischer, D.B.; Bale, T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 2013, 110, 5169–5174. [Google Scholar] [CrossRef] [Green Version]
- Howerton, C.L.; Bale, T.L. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2014, 111, 9639–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, B.M.; O’Donnell, C.M.; Epperson, C.N.; Bale, T.L. Placental H3K27me3 establishes female resilience to prenatal insults. Nat. Commun. 2018, 9, 2555. [Google Scholar] [CrossRef]
- Lebow, M.A.; Schroeder, M.; Tsoory, M.; Holzman-Karniel, D.; Mehta, D.; Ben-Dor, S.; Gil, S.; Bradley, B.; Smith, A.K.; Jovanovic, T. Glucocorticoid-induced leucine zipper ‘quantifies’ stressors and increases male susceptibility to PTSD. Transl. Psychiatry 2019, 9, 178. [Google Scholar] [CrossRef] [Green Version]
- Clifton, V.L.; Cuffe, J.; Moritz, K.M.; Cole, T.J.; Fuller, P.J.; Lu, N.Z.; Kumar, S.; Chong, S.; Saif, Z. Review: The role of multiple placental glucocorticoid receptor isoforms in adapting to the maternal environment and regulating fetal growth. Placenta 2017, 54, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saif, Z.; Hodyl, N.A.; Hobbs, E.; Tuck, A.R.; Butler, M.S.; Osei-Kumah, A.; Clofton, V.L. The human placenta expresses multiple glucocorticoid receptor isoforms that are altered by fetal sex, growth restriction and maternal asthma. Placenta 2014, 35, 260–268. [Google Scholar] [CrossRef]
- Bourke, C.H.; Neigh, G.N. Behavioral effects of chronic adolescent stress are sustained and sexually dimorphic. Horm. Behav. 2011, 60, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Bourke, C.H.; Raees, M.Q.; Malviya, S.; Bradburn, C.A.; Binder, E.B.; Neigh, G.N. Glucocorticoid sensitizers Bag1 and Ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology 2013, 38, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.; Singaravelu, J.; Bhatnagar, S. Enduring and sex-specific effects of adolescent social isolation in rats on adult stress reactivity. Brain. Res. 2010, 1343, 83–92. [Google Scholar] [CrossRef]
- Taylor, S.B.; Taylor, A.R.; Koenig, J.I. The interaction of disrupted type II neuregulin 1 and chronic adolescent stress on adult anxiety- and fear-related behaviors. Neuroscience 2013, 249, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Barha, C.K.; Brummelte, S.; Lieblich, S.E.; Galea, L.A.M. Chronic restraint stress in adolescence differentially influences hypothalamic-pituitary-adrenal axis function and adult hippocampal neurogenesis in male and female rats. Hippocampus 2011, 21, 1216–1227. [Google Scholar] [CrossRef]
- Wulsin, A.C.; Wick-Carlson, D.; Packard, B.A.; Morano, R.; Herman, J.P. Adolescent chronic stress causes hypothalamo-pituitary-adrenocortical hypo-responsiveness and depression-like behavior in adult female rats. Psychoneuroendocrinology 2016, 65, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Cotella, E.M.; Morano, R.L.; Wulsin, A.C.; Martelle, S.M.; Lemen, P.; Fitzgerald, M.; Packard, B.A.; Moloney, R.D.; Herman, J.R. Lasting Impact of Chronic Adolescent Stress and Glucocorticoid Receptor Selective Modulation in Male and Female Rats. Psychoneuroendocrinology 2020, 112, 104490. [Google Scholar] [CrossRef]
- Tottenham, N.; Galván, A. Stress and the adolescent brain: Amygdala-prefrontal cortex circuitry and ventral striatum as developmental targets. Neurosci. Biobehav. Rev. 2016, 70, 217–227. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef]
- Kroon, J.; Pereira, A.M.; Meijer, O.C. Glucocorticoid Sexual Dimorphism in Metabolism: Dissecting the Role of Sex Hormones. Trends. Endocrinol. Metab. 2020, 31, 357–367. [Google Scholar] [CrossRef]
- Woitowich, N.C.; Beery, A.; Woodruff, T. A 10-year follow-up study of sex inclusion in the biological sciences. Elife 2020, 9, e56344. [Google Scholar] [CrossRef]
- Chan, J.C.; Morgan, C.P.; Adrian Leu, N.; Shetty, A.; Cisse, Y.M.; Nugent, B.M.; Morisson, K.E.; Jašarević, E.; Huang, W.; Kanyuch, N.; et al. Reproductive tract extracellular vesicles are sufficient to transmit intergenerational stress and program neurodevelopment. Nat. Commun. 2020, 11, 1499. [Google Scholar] [CrossRef] [Green Version]
- Droste, S.K.; de Groote, L.; Lightman, S.L.; Reul, J.M.H.M.; Linthorst, A.C.E. The ultradian and circadian rhythms of free corticosterone in the brain are not affected by gender: An in vivo microdialysis study in Wistar rats. J. Neuroendocrinol. 2009, 21, 132–140. [Google Scholar] [CrossRef]
- Stroud, L.R.; Salovey, P.; Epel, E.S. Sex differences in stress responses: Social rejection versus achievement stress. Biol. Psychiatry 2002, 52, 318–327. [Google Scholar] [CrossRef]
- Dedovic, K.; Wadiwalla, M.; Engert, V.; Pruessner, J.C. The role of sex and gender socialization in stress reactivity. Dev. Psychol. 2009, 45, 45–55. [Google Scholar] [CrossRef]
- Editorial. Life stresses. Nature 2012, 490, 143. [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moisan, M.-P. Sexual Dimorphism in Glucocorticoid Stress Response. Int. J. Mol. Sci. 2021, 22, 3139. https://doi.org/10.3390/ijms22063139
Moisan M-P. Sexual Dimorphism in Glucocorticoid Stress Response. International Journal of Molecular Sciences. 2021; 22(6):3139. https://doi.org/10.3390/ijms22063139
Chicago/Turabian StyleMoisan, Marie-Pierre. 2021. "Sexual Dimorphism in Glucocorticoid Stress Response" International Journal of Molecular Sciences 22, no. 6: 3139. https://doi.org/10.3390/ijms22063139
APA StyleMoisan, M. -P. (2021). Sexual Dimorphism in Glucocorticoid Stress Response. International Journal of Molecular Sciences, 22(6), 3139. https://doi.org/10.3390/ijms22063139