
Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
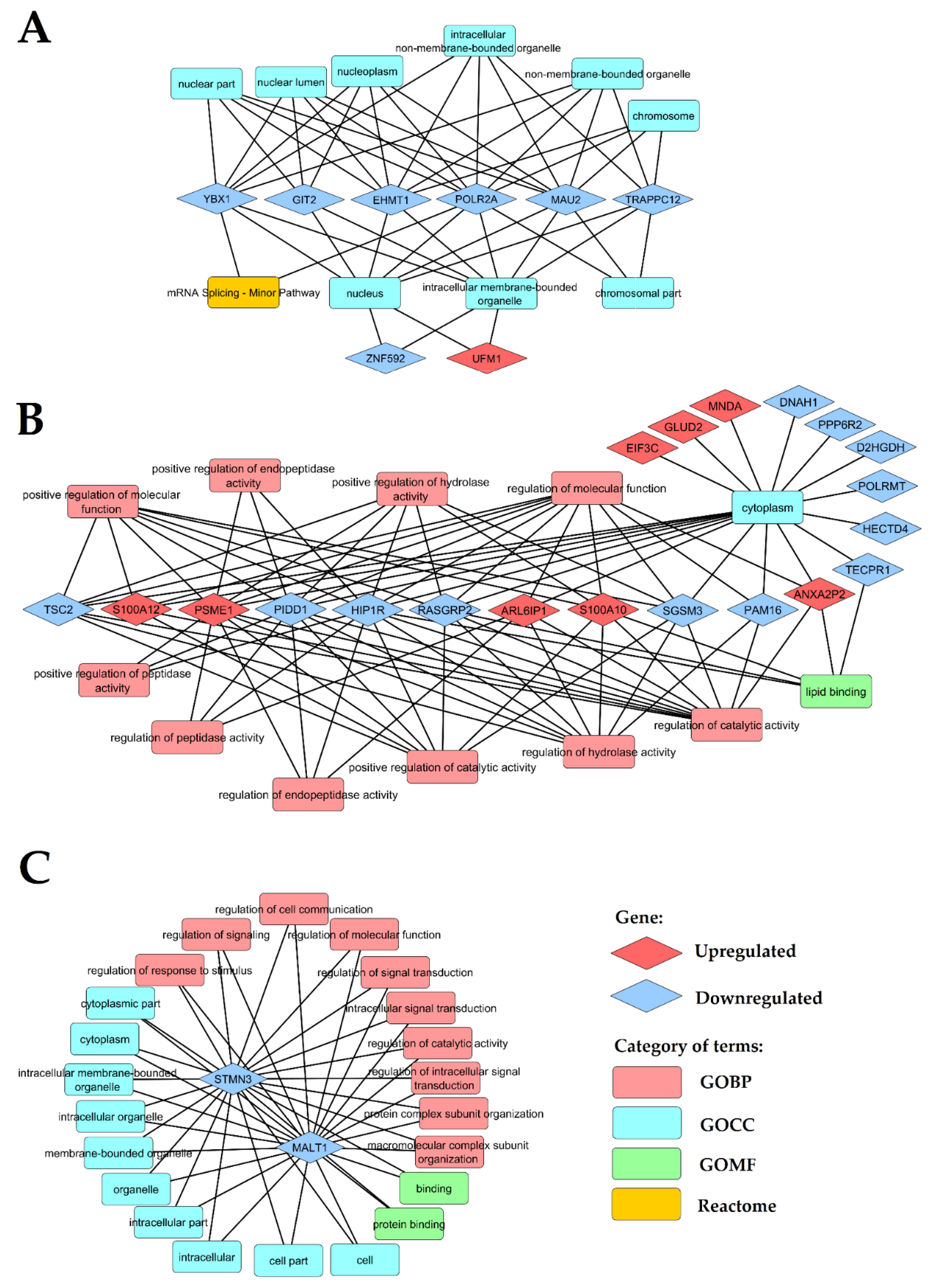
2. Results
2.1. Study Group Characteristics
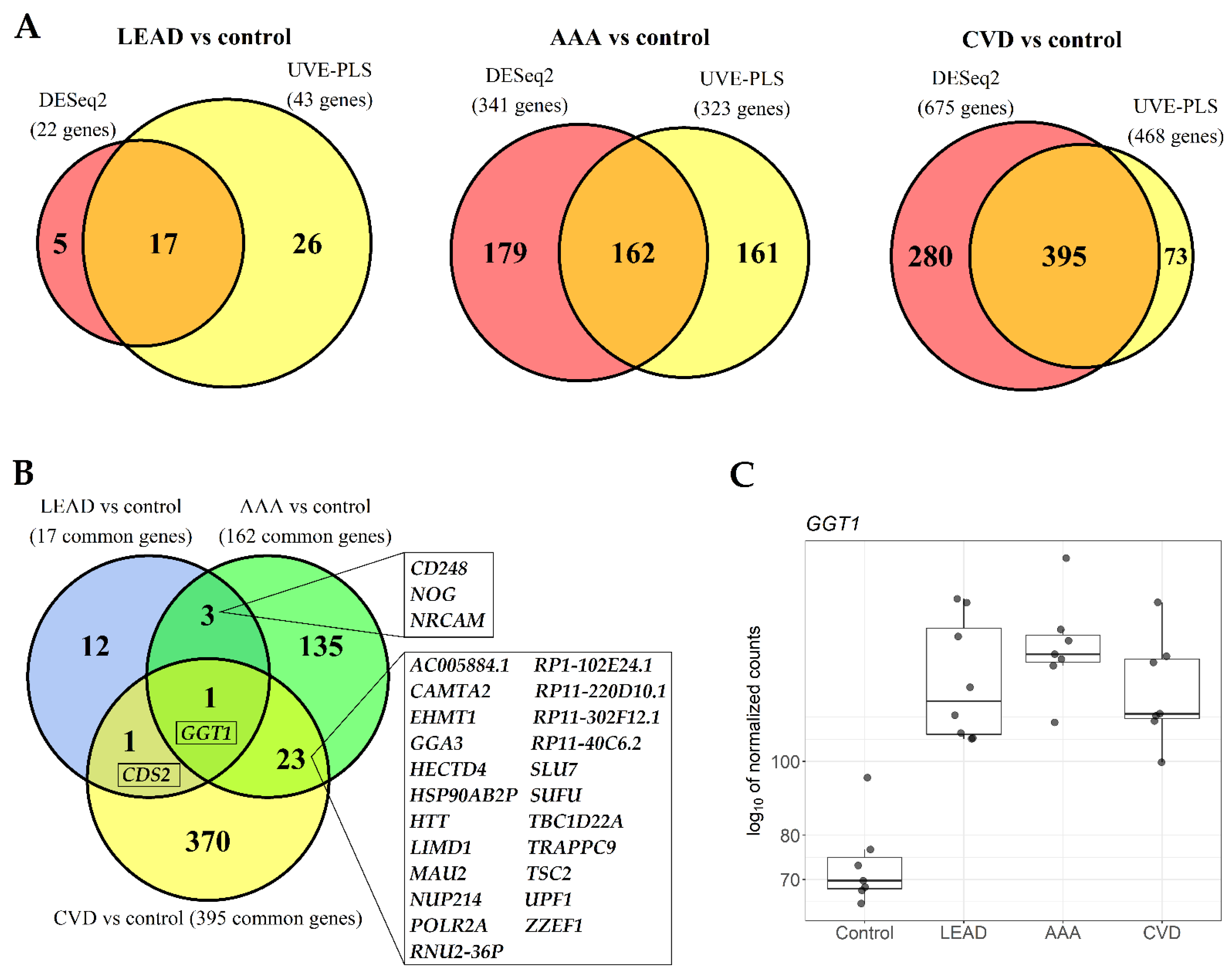
2.2. The Comparison of Differentially Expressed Genes in PBMCs of LEAD, AAA and CVD Subjects in Relation to Healthy Controls
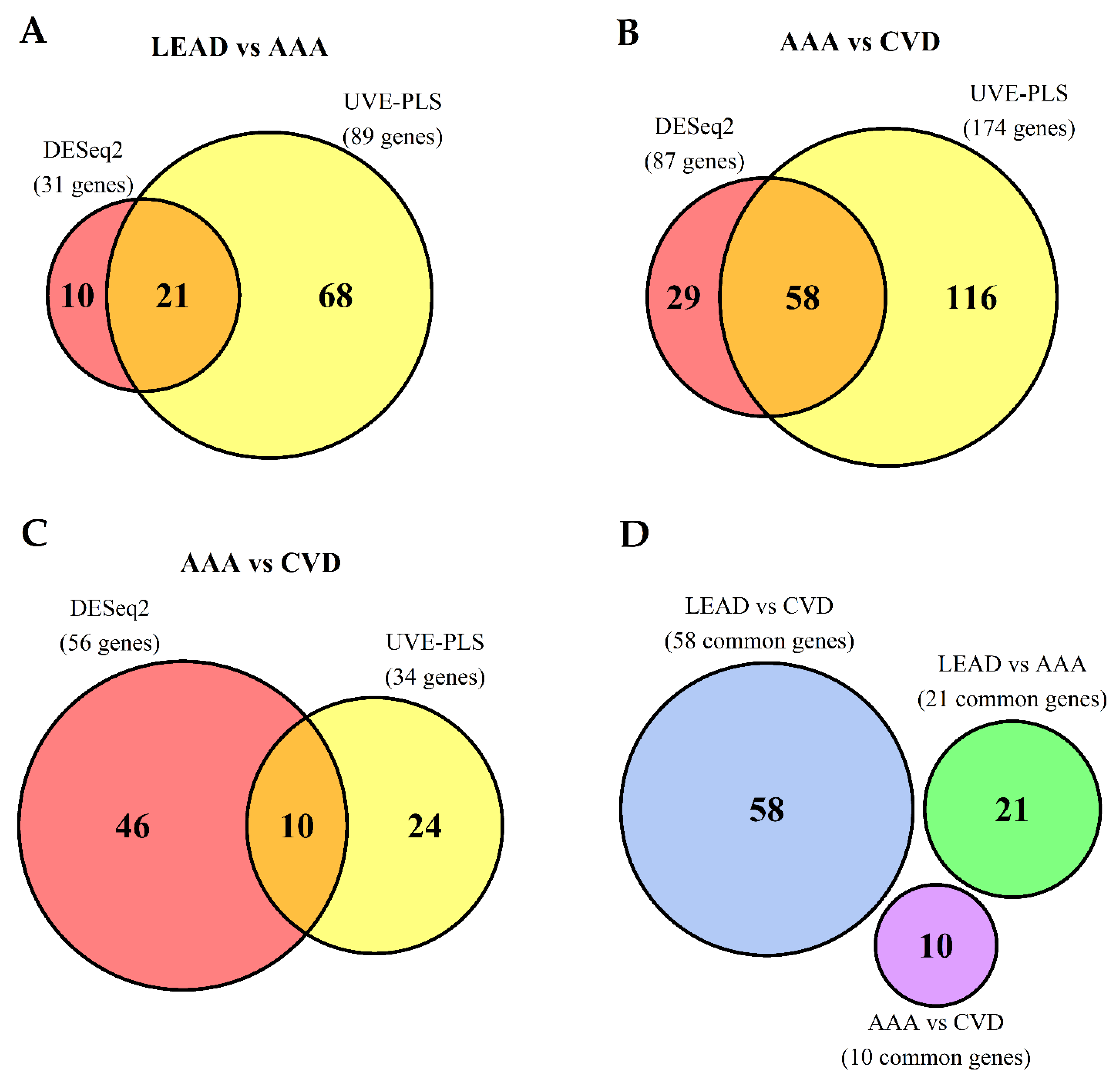
2.3. The Comparison of Differentially Expressed Genes in PBMCs of LEAD, AAA and CVD Subjects after Direct, Pairwise Comparisons
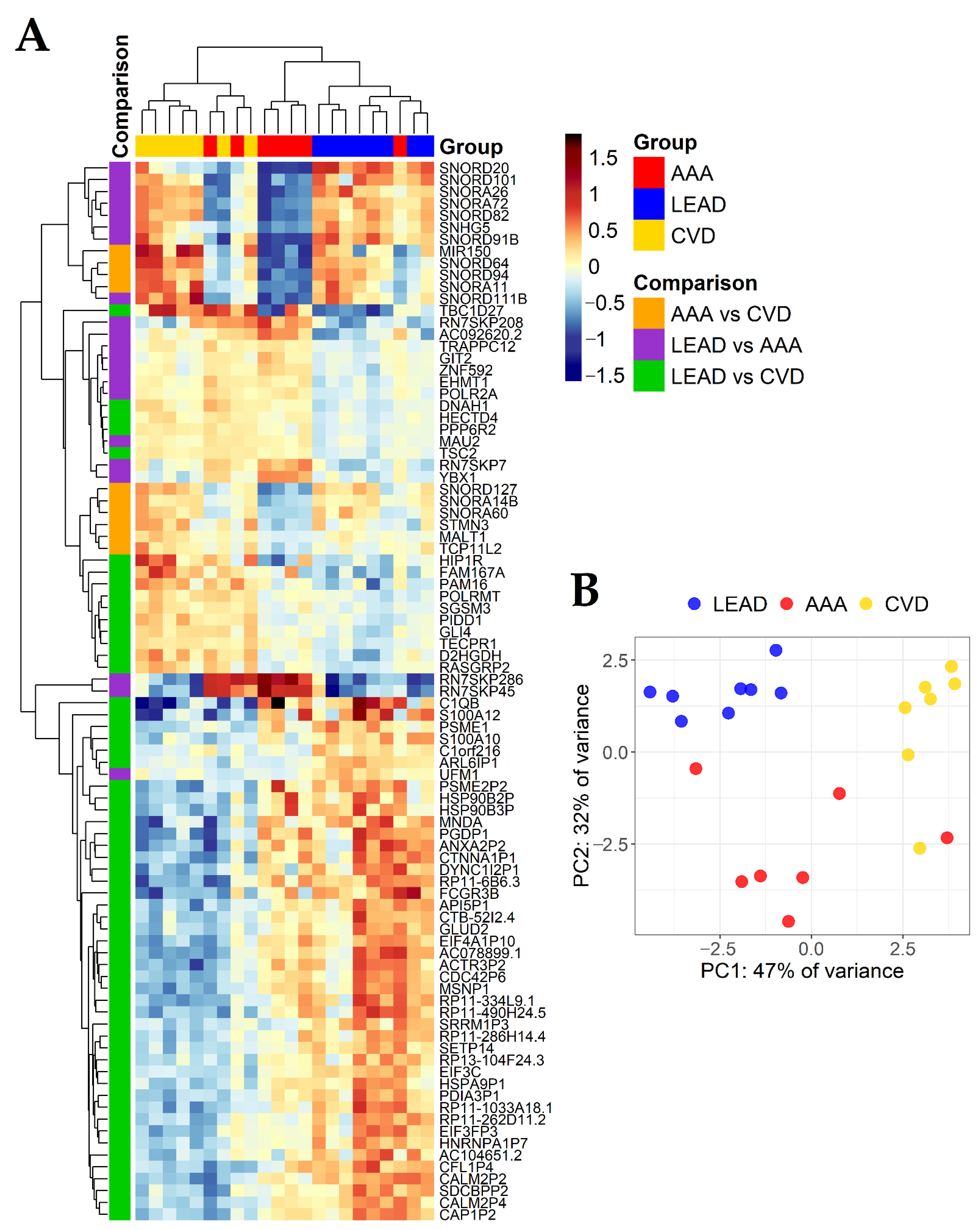
2.4. Identification of Relationships between the Study Group Characteristics and Expression of Genes Found as Unique for LEAD vs. AAA, LEAD vs. CVD and AAA vs. CVD Comparisons
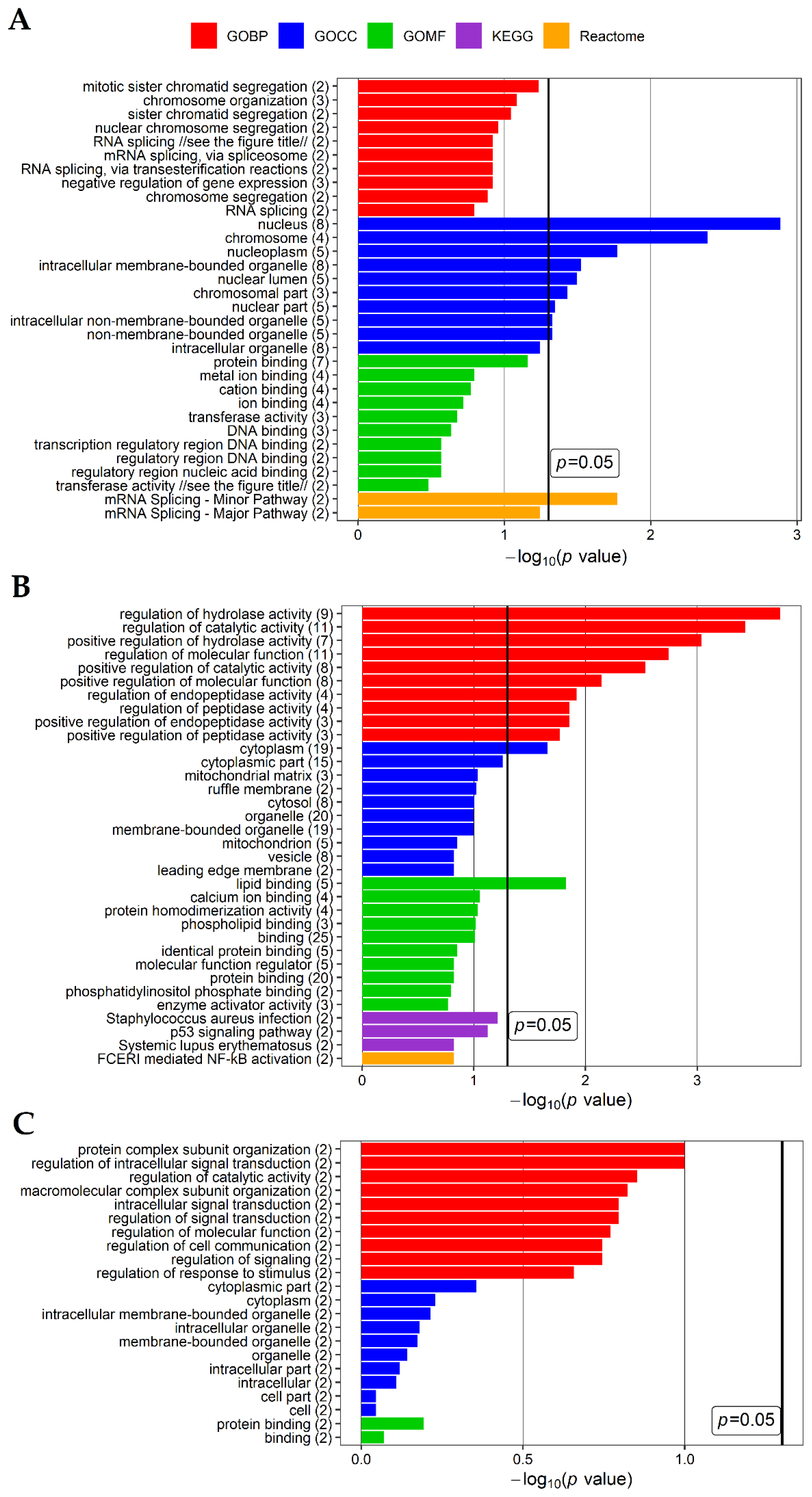
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. Gene Expression Datasets
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | Abdominal Aortic Aneurysm |
| AMPK | AMP-activated protein kinase |
| BMI | Body Mass Index |
| CVD | Chronic Venous Disease |
| DAVID | Database for Annotation, Visualization and Integrated Discovery |
| GOBP | Gene Ontology Biological Processing |
| GOCC | Gene Ontology Cellular Compartment |
| GOMF | Gene Ontology Molecular Function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LEAD | Lower Extremities Arterial Disease |
| miRNA | microRNA |
| PAD | Peripheral Arterial Disease |
| PBMCs | Peripheral Blood Mononuclear Cells |
| PCA | Principal Component Analysis |
| PLS | Partial Least Squares |
| ROC | Receiver Operating Characteristics |
| ROC-AUC | Area under ROC curve |
| snoRNA | Small nucleolar RNA |
| UVE-PLS | Uninformative Variable Elimination by Partial Least Squares |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Parameter | |
|---|---|---|
| LEAD (n = 8) | Indication for treatment: | |
| Rutherford category 2 | 6 (75%) | |
| Rutherford category 3 | 2 (25%) | |
| Initial claudication distance (m) | 143.75 ± 26.69 (100–180) 1 | |
| Ankle-brachial index | 0.658 ± 0.045 (0.59–0.72) 1 | |
| Length of occlusion (cm) | 13.63 ± 5.15 (7–22) 1 | |
| Plaque localization: | ||
| Iliac artery | 1 (12.5%) | |
| Femoral artery | 6 (75%) | |
| Iliac and femoral artery | 1 (12.5%) | |
| AAA (n = 7) | Abdominal aneurysm measurements: | |
| Maximum aneurysm diameter (cm) | 6.371 ± 0.419 (5.8–7.0) 1 | |
| Thrombus volume (cm3) | 10.821 ± 2.605 (6.3–14.7) 1 | |
| Aneurysm neck length (cm) | 0.971 ± 0.198 (0.7–1.2) 1 | |
| CVD (n = 7) | Signs and symptoms: | |
| Pain | 2 (28.6%) | |
| Ankle-brachial index | 0.974 ± 0.016 (0.95–0.99) 1 | |
| Extended anatomical classification: | ||
| Great saphenous vein (above knee) | 3 (42.8%) | |
| Great saphenous vein (below knee) | 2 (28.6%) | |
| Small saphenous vein | 2 (28.6%) | |
| Medication: | ||
| Micronized diosmin | 3 (42.98) | |
| Preparation with vitamin C, hesperidin and Ruscus aculeatus extract | 2 (28.6%) | |
| Both medications | 2 (28.6%) | |
Appendix B
| Comparison | DESeq2 | UVE-PLS | Number of Genes Common for Sets of Genes Selected from DESeq2 (p < 0.001) and from UVE-PLS (Reliability Score ≥8) | |||
|---|---|---|---|---|---|---|
| Number of All Differentially Expressed Genes | Number of Differentially Expressed Genes with p < 0.05 | NUMBER of Differentially Expressed Genes with p < 0.001 | Number of PLS Components/Iterations | Number of Informative Genes with Reliability Score ≥8 | ||
| LEAD vs. AAA | 21,460 | 544 | 31 | 3/1000 | 89 | 21 |
| LEAD vs. CVD | 21,460 | 1603 | 87 | 3/1000 | 174 | 58 |
| AAA vs. CVD | 20,550 | 685 | 56 | 2/1000 | 34 | 10 |
References
- Fuster, V.; Kelly, B.B. Promoting Cardiovascular Health in the Developing World: A Critical Challenge to Acheive Global Health; The National Academies Press: Washington, DC, USA, 2010; ISBN 9780309147743. [Google Scholar]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the global burden of cardiovascular disease, part 1: The epidemiology and risk factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Kaptoge, S.; Pennells, L.; De Bacquer, D.; Cooney, M.T.; Kavousi, M.; Stevens, G.; Riley, L.M.; Savin, S.; Khan, T.; Altay, S.; et al. World Health Organization cardiovascular disease risk charts: Revised models to estimate risk in 21 global regions. Lancet Glob. Health 2019, 7, e1332–e1345. [Google Scholar] [CrossRef] [Green Version]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef] [Green Version]
- Hamburg, N.M.; Creager, M.A. Pathophysiology of intermittent claudication in peripheral artery disease. Circ. J. 2017, 81, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Rudan, D.; Zhu, Y.; Fowkes, F.J.I.; Rahimi, K.; Fowkes, F.G.R.; Rudan, I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: An updated systematic review and analysis. Lancet Glob. Health 2019, 7, e1020–e1030. [Google Scholar] [CrossRef] [Green Version]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Nehler, M.R.; Duval, S.; Diao, L.; Annex, B.H.; Hiatt, W.R.; Rogers, K.; Zakharyan, A.; Hirsch, A.T. Epidemiology of peripheral arterial disease and critical limb ischemia in an insured national population. J. Vasc. Surg. 2014, 60, 686–695.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaikof, E.L.; Dalman, R.L.; Eskandari, M.K.; Jackson, B.M.; Lee, W.A.; Mansour, M.A.; Mastracci, T.M.; Mell, M.; Murad, M.H.; Nguyen, L.L.; et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J. Vasc. Surg. 2018, 67, 2–77.e2. [Google Scholar] [CrossRef] [Green Version]
- Lattanzi, S. Abdominal aortic aneurysms: Pathophysiology and clinical issues. J. Intern. Med. 2020, 288, 376–378. [Google Scholar] [CrossRef]
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.J.; Gelijns, A.C.; Greco, G. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.; Ghoorah, K.; Kunadian, V. Abdominal aortic aneurysms and risk factors for adverse events. Cardiol. Rev. 2016, 24, 88–93. [Google Scholar] [CrossRef]
- Golledge, J.; Kuivaniemi, H. Genetics of abdominal aortic aneurysm. Curr. Opin. Cardiol. 2013, 28, 290–296. [Google Scholar] [CrossRef]
- Bown, M.J. Genomic insights into abdominal aortic aneurysms. Ann. R. Coll. Surg. Engl. 2014, 96, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Alamoudi, A.O.; Haque, S.; Srinivasan, S.; Mital, D.P. Diagnostic efficacy value in terms of sensitivity and specificity of imaging modalities in detecting the abdominal aortic aneurysm: A systematic review. Int. J. Med. Eng. Inform. 2015, 7, 15–35. [Google Scholar] [CrossRef]
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Abbasoglu Ozgoren, A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Krishna, S.M.; Morton, S.K.; Li, J.; Golledge, J. Risk factors and mouse models of abdominal aortic aneurysm rupture. Int. J. Mol. Sci. 2020, 21, 7250. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, R.T.; Raffetto, J.D. Chronic venous insufficiency. Circulation 2014, 130, 333–346. [Google Scholar] [CrossRef]
- Ligi, D.; Croce, L.; Mannello, F. Chronic venous disorders: The dangerous, the good, and the diverse. Int. J. Mol. Sci. 2018, 19, 2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaides, A.; Kakkos, S.; Baekgaard, N.; Comerota, A.; De Maeseneer, M.; Eklof, B.; Giannoukas, A.; Lugli, M.; Maleti, O.; Mansilha, A.; et al. Management of chronic venous disorders of the lower limbs: Guidelines according to scientific evidence part II (Chapters 9–18). Int. Angiol. 2020, 39, 175–230. [Google Scholar] [CrossRef] [PubMed]
- Mansilha, A.; Sousa, J. Pathophysiological mechanisms of chronic venous disease and implications for venoactive drug therapy. Int. J. Mol. Sci. 2018, 19, 1669. [Google Scholar] [CrossRef] [Green Version]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef]
- Vuylsteke, M.E.; Colman, R.; Thomis, S.; Guillaume, G.; Van Quickenborne, D.; Staelens, I. An Epidemiological Survey of Venous Disease Among General Practitioner Attendees in Different Geographical Regions on the Globe: The Final Results of the Vein Consult Program. Angiology 2018, 69, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.S.; Bradbury, A.W.; Kolh, P.; White, J.V.; Dick, F.; Fitridge, R.; Mills, J.L.; Ricco, J.B.; Suresh, K.R.; Murad, M.H.; et al. Global Vascular Guidelines on the Management of Chronic Limb-Threatening Ischemia. Eur. J. Vasc. Endovasc. Surg. 2019, 58, S1–S109.e33. [Google Scholar] [CrossRef] [Green Version]
- Wittens, C.; Davies, A.H.; Bækgaard, N.; Broholm, R.; Cavezzi, A.; Chastanet, S.; De Wolf, M.; Eggen, C.; Giannoukas, A.; Gohel, M.; et al. Editor’s choice—Management of chronic venous disease: Clinical practice guidelines of the European Society for Vascular Surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2015, 49, 678–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Sorescu, D.; Lassègue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Kim, T.M.; Malik, A.B. Transcriptional regulation of endothelial cell and vascular development. Circ. Res. 2013, 112, 1380–1400. [Google Scholar] [CrossRef]
- Harris, B.H.L.; Barberis, A.; West, C.M.L.; Buffa, F.M. Gene Expression Signatures as Biomarkers of Tumour Hypoxia. Clin. Oncol. 2015, 27, 547–560. [Google Scholar] [CrossRef]
- Jeong, H.W.; Hernández-Rodríguez, B.; Kim, J.M.; Kim, K.P.; Enriquez-Gasca, R.; Yoon, J.; Adams, S.; Schöler, H.R.; Vaquerizas, J.M.; Adams, R.H. Transcriptional regulation of endothelial cell behavior during sprouting angiogenesis. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Chen, H.H.; Stewart, A.F.R. Transcriptomic Signature of Atherosclerosis in the Peripheral Blood: Fact or Fiction? Curr. Atheroscler. Rep. 2016, 18, 77. [Google Scholar] [CrossRef]
- Hueso, M.; Mallén, A.; Casas, Á.; Guiteras, J.; Sbraga, F.; Blasco-Lucas, A.; Lloberas, N.; Torras, J.; Cruzado, J.M.; Navarro, E. Integrated miRNA/mRNA counter-expression analysis highlights oxidative stress-related genes CCR7 and FOXO1 as blood markers of coronary arterial disease. Int. J. Mol. Sci. 2020, 21, 1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.M.; Jeong, K.; Steve Lim, S.T. FAK family kinases in vascular diseases. Int. J. Mol. Sci. 2020, 21, 3630. [Google Scholar] [CrossRef]
- Kullo, I.J.; Leeper, N.J. The Genetic Basis of Peripheral Arterial Disease: Current Knowledge, Challenges, and Future Directions. Circ. Res. 2015, 116, 1551–1560. [Google Scholar] [CrossRef] [Green Version]
- Belkin, N.; Damrauer, S.M. Peripheral Arterial Disease Genetics: Progress to Date and Challenges Ahead. Curr. Cardiol. Rep. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Bogucka-Kocka, A.; Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Komsta, Ł.; Kołodziej, P.; Zubilewicz, T.; Feldo, M.; et al. Dysregulation of MicroRNA Regulatory Network in Lower Extremities Arterial Disease. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Pahl, M.C.; Erdman, R.; Kuivaniemi, H.; Lillvis, J.H.; Elmore, J.R.; Tromp, G. Transcriptional (ChIP-Chip) analysis of ELF1, ETS2, RUNX1 and STAT5 in human abdominal aortic aneurysm. Int. J. Mol. Sci. 2015, 16, 11229–11258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, H.Z.; Sylvius, N.; Salem, M.K.; Wild, J.B.; Dattani, N.; Sayers, R.D.; Bown, M.J. Microarray-based Gene Expression Profiling of Abdominal Aortic Aneurysm. Eur. J. Vasc. Endovasc. Surg. 2016, 52, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cai, Z.; Wu, K.; Hu, Y.; Liu, L.; Liao, M. Identification of key microRNAs and genes associated with abdominal aortic aneurysm based on the gene expression profile. Exp. Physiol. 2020, 105, 160–173. [Google Scholar] [CrossRef]
- Gurung, R.; Choong, A.M.; Woo, C.C.; Foo, R.; Sorokin, V. Genetic and epigenetic mechanisms underlying vascular smooth muscle cell phenotypic modulation in abdominal aortic aneurysm. Int. J. Mol. Sci. 2020, 21, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Komsta, Ł.; Kołodziej, P.; Chmiel, P.; Zubilewicz, T.; Feldo, M.; et al. Dysregulation of microRNA Modulatory Network in Abdominal Aortic Aneurysm. J. Clin. Med. 2020, 9, 1974. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.N.; Shortell, C.K. Genomics of varicose veins and chronic venous insufficiency. Semin. Vasc. Surg. 2013, 26, 2–13. [Google Scholar] [CrossRef]
- Grant, Y.; Onida, S.; Davies, A. Genetics in chronic venous disease. Phlebology 2017, 32, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Serralheiro, P.; Novais, A.; Cairrão, E.; Maia, C.; Costa Almeida, C.M.; Verde, I. Variability of MMP/TIMP and TGF-β1 receptors throughout the clinical progression of chronic venous disease. Int. J. Mol. Sci. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Komsta, Ł.; Kołodziej, P.; Chmiel, P.; Zubilewicz, T.; Feldo, M.; et al. Dysregulations of MicroRNA and Gene Expression in Chronic Venous Disease. J. Clin. Med. 2020, 9, 1251. [Google Scholar] [CrossRef]
- Shankar, A.; Li, J.; Klein, B.E.K.; Javier Nieto, F.; Klein, R. Serum gamma-glutamyltransferase level and peripheral arterial disease. Atherosclerosis 2008, 199, 102–109. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kang, Y.M.; Hwang, J.Y.; Kim, E.H.; Yang, D.H.; Kang, J.W.; Park, J.Y.; Lee, W.J.; Kim, H.K.; Jung, C.H. Association between serum gamma-glutamyltransferase and the progression of coronary artery calcification. Atherosclerosis 2015, 243, 300–306. [Google Scholar] [CrossRef]
- Kim, Y.G.; Park, G.M.; Lee, S.B.; Yang, D.H.; Kang, J.W.; Lim, T.H.; Kim, H.K.; Choe, J.; Lee, S.W.; Kim, Y.H. Association of gamma-glutamyl transferase with subclinical coronary atherosclerosis and cardiac outcomes in non-alcoholics. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Lippi, G.; Salvagno, G.L.; Targher, G.; Montagnana, M.; Guidi, G.C. Plasma γ-glutamyl transferase activity predicts homocysteine concentration in a large cohort of unselected outpatients. Intern. Med. 2008, 47, 705–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenk, G.M.; Tromp, G.; Weinsheimer, S.; Gatalica, Z.; Berguer, R.; Kuivaniemi, H. Whole genome expression profiling reveals a significant role for immune function in human abdominal aortic aneurysms. BMC Genom. 2007, 8, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Forman, H.J.; Choi, J. Γ-Glutamyl Transpeptidase in Glutathione Biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Lukic, A.; Ji, J.; Idborg, H.; Samuelsson, B.; Palmberg, L.; Gabrielsson, S.; Rådmark, O. Pulmonary epithelial cancer cells and their exosomes metabolize myeloid cell-derived leukotriene C4to leukotriene D4. J. Lipid Res. 2016, 57, 1659–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondeti, V.; Al-Azzam, N.; Duah, E.; Thodeti, C.K.; Boyce, J.A.; Paruchuri, S. Leukotriene D4 and prostaglandin E2 signals synergize and potentiate vascular inflammation in a mast cell-dependent manner through cysteinyl leukotriene receptor 1 and E-prostanoid receptor 3. J. Allergy Clin. Immunol. 2016, 137, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Liu, B.; Liu, L.; Zhang, E.-L.; Sun, B.; Xu, G.; Chen, J.; Gao, Y. Arachidonic acid metabolism pathway is not only dominant in metabolic modulation but associated with phenotypic variation after acute hypoxia exposure. Front. Physiol. 2018, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13, R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyana-Sundaram, S.; Kumar-Sinha, C.; Shankar, S.; Robinson, D.R.; Wu, Y.M.; Cao, X.; Asangani, I.A.; Kothari, V.; Prensner, J.R.; Lonigro, R.J.; et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell 2012, 149, 1622–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalenko, T.F.; Patrushev, L.I. Pseudogenes as Functionally Significant Elements of the Genome. Biochemistry 2018, 83, 1332–1349. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V. Long Non-coding RNAs in Human Disease. Curr. Top. Microbiol. Immunol. 2016, 394. [Google Scholar] [CrossRef]
- Henras, A.K.; Plisson-Chastang, C.; Humbert, O.; Romeo, Y.; Henry, Y. Synthesis, Function, and Heterogeneity of snoRNA-Guided Posttranscriptional Nucleoside Modifications in Eukaryotic Ribosomal RNAs. Enzymes 2017, 41, 169–213. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shi, J.; Cao, J.; Dong, B.; Guan, W. Latest advances of long non-coding RNA SNHG5 in human cancers. Onco Targets Ther. 2020, 13, 6393–6403. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liao, X.; Zhu, X.; Lv, P.; Li, R. Identification of potential prognostic small nucleolar RNA biomarkers for predicting overall survival in patients with sarcoma. Cancer Med. 2020, 9, 7018–7033. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, R.; Gao, M.; Zhao, Y.; Lv, X.; Zhu, W.; Han, L.; Su, P.; Fan, Y.; Yan, Y.; et al. SNORA72 Activates the Notch1/c-Myc Pathway to Promote Stemness Transformation of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; Macarthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaresma, A.J.C.; Bugai, A.; Barboric, M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016, 44, 7527–7539. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, M.S.; Ding, Y.; Rennie, W.A.; Lane, M.J.; Hanes, S.D. The bin3 RNA methyltransferase targets 7SK RNA to control transcription and translation. Wiley Interdiscip. Rev. RNA 2012, 3, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Singh, N.; Li, H. Essential role of Ufm1 conjugation in the hematopoietic system. Exp. Hematol. 2016, 44, 442–446. [Google Scholar] [CrossRef] [Green Version]
- Renneville, A.; Van Galen, P.; Canver, M.C.; McConkey, M.; Krill-Burger, J.M.; Dorfman, D.M.; Holson, E.B.; Bernstein, B.E.; Orkin, S.H.; Bauer, D.E.; et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood 2015, 126, 1930–1939. [Google Scholar] [CrossRef] [Green Version]
- Tantawy, A.A.G.; Adly, A.A.M.; El Maaty, M.G.A.; Amin, S.A.G. Subclinical atherosclerosis in young β-thalassemia major patients. Hemoglobin 2009, 33, 463–474. [Google Scholar] [CrossRef]
- Salton, M.; Voss, T.C.; Misteli, T. Identification by high-throughput imaging of the histone methyltransferase EHMT2 AS an epigenetic regulator of VEGFA alternative splicing. Nucleic Acids Res. 2014, 42, 13662–13672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.M.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Zhu, N.; Li, L.; Zhang, Y.; Chen, Y.; Zhang, J.; Li, J.; Gao, C. Y-box binding protein 1 regulates ox-LDL mediated inflammatory responses and lipid uptake in macrophages. Free Radic. Biol. Med. 2019, 141, 10–20. [Google Scholar] [CrossRef]
- Van Gastel, J.; Boddaert, J.; Jushaj, A.; Premont, R.T.; Luttrell, L.M.; Janssens, J.; Martin, B.; Maudsley, S. GIT2—A keystone in ageing and age-related disease. Ageing Res. Rev. 2018, 43, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, A.H.; Jia, Z.W.; Pu, K.; Chen, K.Y.; Guo, H. Genome-wide DNA methylation patterns in coronary heart disease. Herz 2018, 43, 656–662. [Google Scholar] [CrossRef]
- Oesterle, A.; Hofmann Bowman, M.A. S100A12 and the S100/Calgranulins: Emerging Biomarkers for Atherosclerosis and Possibly Therapeutic Targets. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2496–2507. [Google Scholar] [CrossRef] [Green Version]
- Joo, H.C.; Choi, J.W.; Moon, H.; Lee, C.Y.; Yoo, K.J.; Kim, S.W.; Hwang, K.C. Protective effects of kenpaullone on cardiomyocytes following H2O2-induced oxidative stress are attributed to inhibition of connexin 43 degradation by SGSM3. Biochem. Biophys. Res. Commun. 2018, 499, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef]
- Ghosh, R.; Vinod, V.; Symons, J.D.; Boudina, S. Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging. Cells 2020, 9, 933. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, L.; Blanchard, S.; Rama, S.; Beier, V.; Kaufmann, A.; Wollert, T. TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Canault, M.; Alessi, M.C. RasGRP2 structure, function and genetic variants in platelet pathophysiology. Int. J. Mol. Sci. 2020, 21, 1075. [Google Scholar] [CrossRef] [Green Version]
- Torres-Do Rego, A.; Barrientos, M.; Ortega-Hernández, A.; Modrego, J.; Gómez-Gordo, R.; Álvarez-Sala, L.A.; Cachofeiro, V.; Gómez-Garre, D. Identification of a Plasma Microrna Signature as Biomarker of Subaneurysmal Aortic Dilation in Patients with High Cardiovascular Risk. J. Clin. Med. 2020, 9, 2783. [Google Scholar] [CrossRef]
- Luo, X.Y.; Zhu, X.Q.; Li, Y.; Wang, X.B.; Yin, W.; Ge, Y.S.; Ji, W.M. MicroRNA-150 restores endothelial cell function and attenuates vascular remodeling by targeting PTX3 through the NF-κB signaling pathway in mice with acute coronary syndrome. Cell Biol. Int. 2018, 42, 1170–1181. [Google Scholar] [CrossRef]
- Ruland, J.; Hartjes, L. CARD–BCL-10–MALT1 signalling in protective and pathological immunity. Nat. Rev. Immunol. 2019, 19, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Selvin, E.; Köttgen, A.; Coresh, J. Kidney function estimated from serum creatinine and cystatin C and peripheral arterial disease in NHANES 1999-2002. Eur. Heart J. 2009, 30, 1918–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, M.M.; Pai, J.K.; Bertoia, M.L.; Gansevoort, R.T.; Bakker, S.J.L.; Cooke, J.P.; Rimm, E.B.; Mukamal, K.J. β2-Microglobulin, cystatin C, and creatinine and risk of symptomatic peripheral artery disease. J. Am. Heart Assoc. 2014, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourrier, M.; Ferguson, T.W.; Embil, J.M.; Rigatto, C.; Komenda, P.; Tangri, N. Peripheral Artery Disease: Its Adverse Consequences With and Without CKD. Am. J. Kidney Dis. 2020, 75, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Xin, G.; Chen, R.; Zhang, X. Candidate gene and mechanism investigations in congenital obstructive nephropathy based on bioinformatics analysis. Mol. Med. Rep. 2018, 18, 2651–2660. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; He, H.; Tan, L.; Wang, L.; Su, Z.; Liu, Y.; Zhu, H.; Zhang, M.; Hou, F.F.; Li, A. Proteomic and phosphoproteomic analysis of renal cortex in a salt-load rat model of advanced kidney damage. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Centner, V.; Massart, D.L.; De Noord, O.E.; De Jong, S.; Vandeginste, B.M.; Sterna, C. Elimination of Uninformative Variables for Multivariate Calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef]
- Mehmood, T.; Liland, K.H.; Snipen, L.; Sæbø, S. A review of variable selection methods in Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 118, 62–69. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






| Characteristic | LEAD (n = 8) | AAA (n = 7) | CVD (n = 7) | p1 |
|---|---|---|---|---|
| Age | 62 ± 7.82 2 | 66.3 ± 4.03 2 | 41.3 ± 4.03 2 | 8.273 × 10−4 4 |
| 48–71 3 | 59–71 3 | 35–47 3 | ||
| Gender males/females | 6 (75%)/2 (25%) | 6 (85.7%)/ | 3 (42.9%)/ | 0.275 5 |
| 1 (14.3%) | 4 (57.1%) | |||
| Body mass index (BMI) | 28.25 ± 2.07 2 | 27.23 ± 2.76 2 | 23.36 ± 1.94 2 | 6.272 × 10−3 4 |
| 25.5–31.2 3 | 23.66–30.85 3 | 20.94–25.83 3 | ||
| Risk factors and cardiovascular comorbidities | ||||
| Smoking never/former/current | 0 (0%)/6 (75%)/ | 3 (42.9%)/ | 5 (71.4%)/0 (0%)/ | 0.011 5 |
| 2 (25%) | 2 (28.6%)/2 (28.6%) | 2 (28.6%) | ||
| Diabetes type 2 | 3 (37.5%) | 2 (28.6%) | 0 (0%) | 0.300 5 |
| Hypertension | 7 (87.5%) | 5 (71.4%) | 0 (0%) | 9.418 × 10−4 5 |
| Coronary artery disease (CAD) | 2 (25%) | 1 (14.3%) | 0 (0%) | 0.746 5 |
| Myocardial infarction | 2 (25%) | 1 (14.3%) | 0 (0%) | 0.746 5 |
| Stroke/Transient ischemic attack | 0 (0%) | 0 (0%) | 0 (0%) | 1.000 5 |
| Hematological and biochemical blood parameters | ||||
| Red blood cells (M/µl) | 4.81 ± 0.33 2 | 4.96 ± 0.19 2 | 4.93 ± 0.31 2 | 0.630 4 |
| 4.22–5.18 3 | 4.56–5.10 3 | 4.29–5.21 3 | ||
| White blood cells (K/µl) | 5.49 ± 0.69 2 | 5.85 ± 0.75 2 | 5.58 ± 0.50 2 | 0.677 4 |
| 4.79–6.70 3 | 4.89–6.89 3 | 4.67–5.99 3 | ||
| Platelets (K/µl) | 348.5 ± 105.5 2 | 379.43 ± 82.26 2 | 368.14 ± 66.26 2 | 0.430 4 |
| 267–432 3 | 267–501 3 | 295–467 3 | ||
| Hemoglobin (g/dl) | 14.22 ± 0.59 2 | 13.88 ± 0.52 2 | 13.98 ± 0.33 2 | 0.415 4 |
| 13.45–14.80 3 | 13.34–14.60 3 | 13.56–14.60 3 | ||
| Hematocrit (%) | 40.91 ± 1.15 2 | 41.31 ± 1.13 2 | 40.24 ± 2.35 2 | 0.425 4 |
| 38.9–42 3 | 39.9–43 3 | 37.00–44 3 | ||
| Creatinine (mmol/L) | 80.38 ± 11.11 2 | 58.86 ± 11.60 2 | 58.71 ± 8.75 2 | 4.529 × 10−3 4 |
| 59–89 3 | 44–77 3 | 45–67 3 | ||
| Urea (mmol/L) | 4.69 ± 0.70 2 | 4.61 ± 0.47 2 | 4.77 ± 0.98 2 | 0.931 4 |
| 3.70–6.01 3 | 3.89–5.10 3 | 3.78–6.37 3 | ||
| Medication | ||||
| Statins | 7 (87.5%) | 4 (57.1%) | 0 (0%) | 2.818 × 10−3 5 |
| Acetylsalicylic acid | 8 (100%) | 7 (100%) | 0 (0%) | 1.173 × 10−5 5 |
| Clopidogrel | 2 (25%) | 0 (0%) | 0 (0%) | 0.303 5 |
| Beta-adrenergic blockers | 6 (75%) | 5 (71.4%) | 0 (0%) | 8.375 × 10−3 5 |
| Angiotensin-converting enzyme inhibitor | 2 (25%) | 0 (0%) | 0 (0%) | 0.303 5 |
| Ca2+ channel blockers | 3 (37.5%) | 1 (14.3%) | 0 (0%) | 0.270 5 |
| Fibrates | 3 (37.5%) | 1 (14.3%) | 0 (0%) | 0.270 5 |
| Metformin | 1 (12.5%) | 0 (0%) | 0 (0%) | 1.000 5 |
| Gliclazide | 3 (37.5%) | 2 (28.6%) | 0 (0%) | 0.300 5 |
| No. | Gene Symbol | Gene Name | p | Fold Change | PLS Coefficient | ROC-AUC |
|---|---|---|---|---|---|---|
| LEAD vs. AAA—Upregulated Genes | ||||||
| 1. | SNORD20 | small nucleolar RNA, C/D box 20 | 1.712 × 10−7 | 3.338 | 2.746 × 10−3 | 1.000 |
| 2. | SNORA72 | small nucleolar RNA, H/ACA box 72 | 1.205 × 10−4 | 2.103 | 1.551 × 10−3 | 0.964 |
| 3. | SNHG5 | small nucleolar RNA host gene 5 | 1.205 × 10−4 | 1.984 | 1.541 × 10−3 | 1.000 |
| 4. | SNORA26 | small nucleolar RNA, H/ACA box 26 | 1.358 × 10−4 | 2.365 | 1.857 × 10−3 | 1.000 |
| 5. | SNORD82 | small nucleolar RNA, C/D box 82 | 1.489 × 10−4 | 2.206 | 1.721 × 10−3 | 1.000 |
| 6. | UFM1 | ubiquitin fold modifier 1 | 2.157 × 10−4 | 1.391 | 7.038 × 10−4 | 1.000 |
| 7. | SNORD101 | small nucleolar RNA, C/D box 101 | 3.292 × 10−4 | 2.352 | 1.757 × 10−3 | 0.964 |
| 8. | SNORD91B | small nucleolar RNA, C/D box 91B | 6.538 × 10−4 | 2.493 | 1.987 × 10−3 | 1.000 |
| 9. | SNORD111B | small nucleolar RNA, C/D box 111B | 7.373 × 10−4 | 2.337 | 1.910 × 10−3 | 1.000 |
| LEAD vs. AAA—downregulated genes | ||||||
| 10. | POLR2A | RNA polymerase II subunit A | 2.583 × 10−5 | 0.764 | −6.306 × 10−4 | 1.000 |
| 11. | AC092620.2 | Unmatched | 2.583 × 10−5 | 0.401 | −1.694 × 10−3 | 1.000 |
| 12. | EHMT1 | euchromatic histone lysine methyltransferase 1 | 2.854 × 10−5 | 0.744 | −6.428 × 10−4 | 1.000 |
| 13. | TRAPPC12 | trafficking protein particle complex 12 | 1.313 × 10−4 | 0.762 | −5.972 × 10−4 | 0.980 |
| 14. | RN7SKP286 | RN7SK pseudogene 286 | 1.313 × 10−4 | 0.143 | −3.159 × 10−3 | 0.964 |
| 15. | ZNF592 | zinc finger protein 592 | 5.389 × 10−4 | 0.740 | −6.769 × 10−4 | 1.000 |
| 16. | YBX1 | Y-box binding protein 1 | 5.525 × 10−4 | 0.625 | −9.797 × 10−4 | 0.982 |
| 17. | RN7SKP208 | RN7SK pseudogene 208 | 5.525 × 10−4 | 0.292 | −1.571 × 10−3 | 0.982 |
| 18. | RN7SKP45 | RN7SK pseudogene 45 | 5.525 × 10−4 | 0.213 | −2.861 × 10−3 | 0.982 |
| 19. | RN7SKP7 | RN7SK pseudogene 7 | 5.525 × 10−4 | 0.199 | −1.277 × 10−3 | 1.000 |
| 20. | MAU2 | MAU2 sister chromatid cohesion factor | 6.538 × 10−4 | 0.804 | −4.892 × 10−4 | 0.982 |
| 21. | GIT2 | GIT ArfGAP 2 | 9.198 × 10−4 | 0.768 | −5.651 × 10−4 | 1.000 |
| LEAD vs. CVD—upregulated genes | ||||||
| 1. | CALM2P2 | calmodulin 2 pseudogene 2 | 4.927 × 10−6 | 2.622 | 1.572 × 10−3 | 1.000 |
| 2. | RP11-490H24.5 | Unmatched | 9.430 × 10−6 | 3.231 | 1.296 × 10−3 | 1.000 |
| 3. | RP11-334L9.1 | Unmatched | 1.438 × 10−5 | 3.236 | 1.468 × 10−3 | 0.982 |
| 4. | API5P1 | apoptosis inhibitor 5 pseudogene 1 | 3.627 × 10−5 | 2.592 | 1.284 × 10−3 | 1.000 |
| 5. | PDIA3P1 | protein disulfide isomerase family A member 3 pseudogene 1 | 3.627 × 10−5 | 1.968 | 1.090 × 10−3 | 1.000 |
| 6. | ARL6IP1 | ADP ribosylation factor like GTPase 6 interacting protein 1 | 3.627 × 10−5 | 1.540 | 8.133 × 10−4 | 1.000 |
| 7. | RP11-1033A18.1 | Unmatched | 4.570 × 10−5 | 2.266 | 1.376 × 10−3 | 1.000 |
| 8. | EIF4A1P10 | eukaryotic translation initiation factor 4A1 pseudogene 10 | 5.014 × 10−5 | 2.026 | 1.131 × 10−3 | 1.000 |
| 9. | RP11-262D11.2 | Unmatched | 5.014 × 10−5 | 1.913 | 1.072 × 10−3 | 0.946 |
| 10. | S100A10 | S100 calcium binding protein A10 | 5.014 × 10−5 | 1.723 | 9.939 × 10−4 | 1.000 |
| 11. | CFL1P4 | cofilin 1 pseudogene 4 | 5.355 × 10−5 | 2.826 | 1.400 × 10−3 | 1.000 |
| 12. | AC078899.1 | Unmatched | 5.355 × 10−5 | 2.411 | 1.334 × 10−3 | 0.982 |
| 13. | CAP1P2 | CAP1 pseudogene 2 | 7.322 × 10−5 | 2.104 | 1.191 × 10−3 | 1.000 |
| 14. | HNRNPA1P7 | heterogeneous nuclear ribonucleoprotein A1 pseudogene 7 | 7.322 × 10−5 | 1.814 | 1.017 × 10−3 | 1.000 |
| 15. | FCGR3B | Fc fragment of IgG receptor IIIb | 9.228 × 10−5 | 3.135 | 1.917 × 10−3 | 1.000 |
| 16. | CTNNA1P1 | catenin alpha 1 pseudogene 1 | 9.228 × 10−5 | 3.030 | 1.412 × 10−3 | 0.982 |
| 17. | PSME1 | proteasome activator subunit 1 | 9.228 × 10−5 | 1.744 | 1.083 × 10−3 | 1.000 |
| 18. | RP11-6B6.3 | Unmatched | 1.126 × 10−4 | 3.206 | 1.602 × 10−3 | 1.000 |
| 19. | MSNP1 | moesin pseudogene 1 | 1.602 × 10−4 | 2.059 | 1.213 × 10−3 | 1.000 |
| 20. | ACTR3P2 | ACTR3 pseudogene 2 | 1.640 × 10−4 | 2.564 | 1.369 × 10−3 | 1.000 |
| 21. | RP13-104F24.3 | Unmatched | 1.640 × 10−4 | 2.143 | 8.857 × 10−4 | 0.982 |
| 22. | HSP90B3P | heat shock protein 90 beta family member 3, pseudogene | 1.987 × 10−4 | 2.373 | 1.277 × 10−3 | 1.000 |
| 23. | DYNC1I2P1 | dynein cytoplasmic 1 intermediate chain 2 pseudogene 1 | 2.024 × 10−4 | 2.441 | 1.344 × 10−3 | 1.000 |
| 24. | EIF3FP3 | eukaryotic translation initiation factor 3 subunit F pseudogene 3 | 2.996 × 10−4 | 1.976 | 1.059 × 10−3 | 0.964 |
| 25. | C1orf216 | chromosome 1 open reading frame 216 | 3.042 × 10−4 | 1.474 | 6.989 × 10−4 | 0.982 |
| 26. | ANXA2P2 | annexin A2 pseudogene 2 | 3.767 × 10−4 | 2.368 | 1.258 × 10−3 | 1.000 |
| 27. | MNDA | myeloid cell nuclear differentiation antigen | 4.212 × 10−4 | 2.198 | 1.322 × 10−3 | 1.000 |
| 28. | AC104651.2 | Unmatched | 4.292 × 10−4 | 3.349 | 9.316 × 10−4 | 0.946 |
| 29. | PGDP1 | phosphogluconate dehydrogenase pseudogene 1 | 4.292 × 10−4 | 2.653 | 1.284 × 10−3 | 0.982 |
| 30. | PSME2P2 | proteasome activator subunit 2 pseudogene 2 | 4.425 × 10−4 | 2.547 | 1.508 × 10−3 | 1.000 |
| 31. | CDC42P6 | cell division cycle 42 pseudogene 6 | 4.693 × 10−4 | 1.981 | 1.051 × 10−3 | 1.000 |
| 32. | HSP90B2P | heat shock protein 90 beta family member 2, pseudogene | 5.142 × 10−4 | 2.048 | 1.107 × 10−3 | 1.000 |
| 33. | HSPA9P1 | heat shock protein family A (Hsp70) member 9 pseudogene 1 | 5.302 × 10−4 | 1.930 | 8.806 × 10−4 | 1.000 |
| 34. | C1QB | complement C1q B chain | 5.647 × 10−4 | 5.492 | 2.159 × 10−3 | 0.964 |
| 35. | CTB-52I2.4 | Unmatched | 5.855 × 10−4 | 2.077 | 1.013 × 10−3 | 0.982 |
| 36. | RP11-286H14.4 | Unmatched | 5.907 × 10−4 | 1.932 | 9.926 × 10−4 | 1.000 |
| 37. | SETP14 | SET pseudogene 14 | 6.672 × 10−4 | 1.785 | 9.681 × 10−4 | 1.000 |
| 38. | CALM2P4 | calmodulin 2 pseudogene 4 | 6.970 × 10−4 | 2.329 | 1.066 × 10−3 | 1.000 |
| 39. | GLUD2 | glutamate dehydrogenase 2 | 7.874 × 10−4 | 1.870 | 8.908 × 10−4 | 0.982 |
| 40. | EIF3C | eukaryotic translation initiation factor 3 subunit C | 8.845 × 10−4 | 1.670 | 9.814 × 10−4 | 1.000 |
| 41. | SDCBPP2 | syndecan binding protein pseudogene 2 | 9.306 × 10−4 | 2.454 | 1.164 × 10−3 | 1.000 |
| 42. | SRRM1P3 | serine/arginine repetitive matrix 1 pseudogene 3 | 9.306 × 10−4 | 2.044 | 1.077 × 10−3 | 1.000 |
| 43. | S100A12 | S100 calcium binding protein A12 | 9.443 × 10−4 | 2.972 | 1.516 × 10−3 | 0.946 |
| LEAD vs. CVD—downregulated genes | ||||||
| 44. | TSC2 | TSC complex subunit 2 | 3.328 × 10−6 | 0.765 | −5.314 × 10−4 | 1.000 |
| 45. | SGSM3 | small G protein signaling modulator 3 | 5.014 × 10−5 | 0.723 | −5.758 × 10−4 | 1.000 |
| 46. | TECPR1 | tectonin beta-propeller repeat containing 1 | 6.319 × 10−5 | 0.716 | −6.552 × 10−4 | 1.000 |
| 47. | RASGRP2 | RAS guanyl releasing protein 2 | 7.322 × 10−5 | 0.663 | −7.661 × 10−4 | 0.964 |
| 48. | GLI4 | GLI family zinc finger 4 | 1.484 × 10−4 | 0.671 | −6.852 × 10−4 | 1.000 |
| 49. | PPP6R2 | protein phosphatase 6 regulatory subunit 2 | 1.640 × 10−4 | 0.773 | −5.375 × 10−4 | 1.000 |
| 50. | TBC1D27P | TBC1 domain family member 27, pseudogene | 1.806 × 10−4 | 0.220 | −2.347 × 10−3 | 1.000 |
| 51. | D2HGDH | D-2-hydroxyglutarate dehydrogenase | 2.024 × 10−4 | 0.589 | −9.321 × 10−4 | 0.964 |
| 52. | DNAH1 | dynein axonemal heavy chain 1 | 2.532 × 10−4 | 0.727 | −5.605 × 10−4 | 1.000 |
| 53. | PAM16 | presequence translocase associated motor 16 | 3.180 × 10−4 | 0.526 | −1.073 × 10−3 | 0.982 |
| 54. | HIP1R | huntingtin interacting protein 1 related | 3.236 × 10−4 | 0.489 | −1.198 × 10−3 | 1.000 |
| 55. | FAM167A | family with sequence similarity 167 member A | 4.088 × 10−4 | 0.331 | −1.221 × 10−3 | 0.982 |
| 56. | PIDD1 | p53-induced death domain protein 1 | 4.292 × 10−4 | 0.682 | −6.748 × 10−4 | 1.000 |
| 57. | HECTD4 | HECT domain E3 ubiquitin protein ligase 4 | 5.855 × 10−4 | 0.785 | −4.282 × 10−4 | 0.982 |
| 58. | POLRMT | RNA polymerase mitochondrial | 6.453 × 10−4 | 0.710 | −5.682 × 10−4 | 1.000 |
| AAA vs. CVD—downregulated genes | ||||||
| 1. | SNORA11 | small nucleolar RNA, H/ACA box 11 | 2.066 × 10−6 | 0.392 | −1.585 × 10−3 | 0.980 |
| 2. | SNORD64 | small nucleolar RNA, C/D box 64 | 4.692 × 10−6 | 0.354 | −1.471 × 10−3 | 0.959 |
| 3. | MIR150 | microRNA 150 | 2.022 × 10−5 | 0.274 | −1.996 × 10−3 | 0.959 |
| 4. | SNORD94 | small nucleolar RNA, C/D box 94 | 3.480 × 10−5 | 0.441 | −1.229 × 10−3 | 0.939 |
| 5. | MALT1 | MALT1 paracaspase | 1.177 × 10−4 | 0.762 | −4.413 × 10−4 | 1.000 |
| 6. | SNORD127 | small nucleolar RNA, C/D box 127 | 1.519 × 10−4 | 0.550 | −9.698 × 10−4 | 0.959 |
| 7. | SNORA14B | small nucleolar RNA, H/ACA box 14B | 4.364 × 10−4 | 0.672 | −6.430 × 10−4 | 0.959 |
| 8. | STMN3 | stathmin 3 | 4.598 × 10−4 | 0.603 | −8.031 × 10−4 | 0.939 |
| 9. | TCP11L2 | t-complex 11 like 2 | 7.061 × 10−4 | 0.689 | −6.071 × 10−4 | 1.000 |
| 10. | SNORA60 | small nucleolar RNA, H/ACA box 60 | 9.366 × 10−4 | 0.641 | −7.161 × 10−4 | 0.959 |
| Comparison | Age | BMI | Creatinine | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | R | p | Gene Symbol | R | p | Gene Symbol | R | p | |
| LEAD vs. AAA | none | none | POLR2A | −0.65 | 2.91 × 10−3 | ||||
| ZNF592 | −0.62 | 5.25 × 10−3 | |||||||
| TRAPPC12 | −0.60 | 7.01 × 10−3 | |||||||
| LEAD vs. CVD | PSME2P2 | 0.69 | 1.27 × 10−3 | TECPR1 | −0.76 | 2.25 × 10−4 | RP11−262D11.2 | 0.72 | 6.68 × 10−4 |
| FCGR3B | 0.69 | 1.35 × 10−3 | PIDD | −0.75 | 2.97 × 10−4 | SRRM1P3 | 0.70 | 9.06 × 10−4 | |
| API5P1 | 0.67 | 1.83 × 10−3 | PSME1 | 0.67 | 2.01 × 10−3 | SDCBPP2 | 0.70 | 9.66 × 10−4 | |
| ACTR3P2 | 0.65 | 2.80 × 10−3 | D2HGDH | −0.66 | 2.42 × 10−3 | ARL6IP1 | 0.67 | 1.83 × 10−3 | |
| CDC42P6 | 0.64 | 3.80 × 10−3 | HSP90B3P | 0.65 | 3.04 × 10−3 | HNRNPA1P7 | 0.67 | 1.83 × 10−3 | |
| HSP90B2P | 0.63 | 4.24 × 10−3 | PPP6R2 | −0.64 | 3.39 × 10−3 | API5P1 | 0.67 | 1.94 × 10−3 | |
| PIDD1 | −0.62 | 4.70 × 10−3 | EIF3C | 0.62 | 3.41 × 10−3 | AC104651.2 | 0.66 | 2.39 × 10−3 | |
| SGSM3 | −0.62 | 4.79 × 10−3 | HSPA9P1 | 0.60 | 6.72 × 10−3 | EIF3FP3 | 0.65 | 2.85 × 10−3 | |
| CAP1P2 | 0.62 | 4.89 × 10−3 | RP11-286H14.4 | 0.65 | 2.88 × 10−3 | ||||
| RP11-6B6.3 | 0.62 | 5.08 × 10−3 | CTNNA1P1 | 0.64 | 3.25 × 10−3 | ||||
| RP11-490H24.5 | 0.62 | 5.14 × 10−3 | DYNC1I2P1 | 0.64 | 3.54 × 10−3 | ||||
| RP13-104F24.3 | 0.61 | 5.55 × 10−3 | CTB-52I2.4 | 0.62 | 5.49 × 10−3 | ||||
| CTB-52I2.4 | 0.61 | 5.65 × 10−3 | |||||||
| HSP90B3P | 0.61 | 5.77 × 10−3 | |||||||
| CTNNA1P1 | 0.60 | 6.83 × 10−3 | |||||||
| AAA vs. CVD | SNORD64 | −0.68 | 1.40 × 10−3 | none | none | ||||
| STMN3 | −0.66 | 2.20 × 10−3 | |||||||
| MIR150 | −0.65 | 2.99 × 10−3 | |||||||
| MALT1 | −0.63 | 4.39 × 10−3 | |||||||
| Comparison | Hypertension Status | Statins Medication | Acetylsalicylic Acid | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | p | Gene Symbol | p | Gene Symbol | p | Gene Symbol | p | Gene Symbol | p | |
| LEAD vs. AAA | none | none | none | |||||||
| LEAD vs. CVD | GLI4 | 8.09 × 10−3 | FAM167A | 2.05 × 10−2 | FCGR3B | 2.09 × 10−3 | SGSM3 | 5.22 × 10−3 | D2HGDH | 1.09 × 10−2 |
| MNDA | 3.52 × 10−2 | C1orf216 | 4.76 × 10−2 | GLI4 | 2.09 × 10−3 | CDC42P6 | 6.02 × 10−3 | DYNC1I2P1 | 1.09 × 10−2 | |
| HSP90B2P | 2.09 × 10−3 | EIF3C | 6.02 × 10−3 | EIF3FP3 | 1.09 × 10−2 | |||||
| PSME2P2 | 2.09 × 10−3 | EIF4A1P10 | 6.02 × 10−3 | PGDP1 | 1.09 × 10−2 | |||||
| TECPR1 | 2.09 × 10−3 | PSME1 | 6.02 × 10−3 | RASGRP2 | 1.09 × 10−2 | |||||
| HSP90B3P | 2.32 × 10−3 | RP11-6B6.3 | 6.02 × 10−3 | SDCBPP2 | 1.09 × 10−2 | |||||
| PIDD1 | 2.32 × 10−3 | ANXA2P2 | 7.34 × 10−3 | CALM2P4 | 1.38 × 10−2 | |||||
| RP11-490H24.5 | 2.32 × 10−3 | CAP1P2 | 7.34 × 10−3 | FAM167A | 1.38 × 10−2 | |||||
| RP13-104F24.3 | 2.32 × 10−3 | HIP1R | 7.34 × 10−3 | RP11-262D11.2 | 1.38 × 10−2 | |||||
| HSPA9P1 | 3.04 × 10−3 | MSNP1 | 7.34 × 10−3 | TBC1D27P | 1.38 × 10−2 | |||||
| PDIA3P1 | 3.04 × 10−3 | POLRMT | 7.34 × 10−3 | C1orf216 | 1.82 × 10−2 | |||||
| SETP14 | 3.04 × 10−3 | RP11-1033A18.1 | 7.34 × 10−3 | TSC2 | 1.82 × 10−2 | |||||
| ACTR3P2 | 3.91 × 10−3 | CALM2P2 | 9.06 × 10−3 | SRRM1P3 | 3.11 × 10−2 | |||||
| API5P1 | 3.91 × 10−3 | CTB-52I2.4 | 9.06 × 10−3 | AC104651.2 | 3.90 × 10−2 | |||||
| C1QB | 3.91 × 10−3 | CTNNA1P1 | 9.06 × 10−3 | CFL1P4 | 3.90 × 10−2 | |||||
| HNRNPA1P7 | 3.91 × 10−3 | GLUD2 | 9.06 × 10−3 | HECTD4 | 4.76 × 10−2 | |||||
| RP11-286H14.4 | 5.22 × 10−3 | MNDA | 9.06 × 10−3 | PAM16 | 4.76 × 10−2 | |||||
| RP11-334L9.1 | 5.22 × 10−3 | AC078899.1 | 1.09 × 10−2 | S100A10 | 4.76 × 10−2 | |||||
| AAA vs. CVD | none | none | TCP11L2 | 6.02 × 10−3 | STMN3 | 1.38 × 10−2 | MIR150 | 3.11 × 10−2 | ||
| MALT1 | 6.02 × 10−3 | |||||||||
| Direction of Regulation | Gene Type | Gene Symbols | Number in up-/Downregulated Group of Genes | % |
|---|---|---|---|---|
| LEAD vs. AAA | ||||
| up | snoRNA | SNORA26, SNORA72, SNORD101, SNORD111B, SNORD20, SNORD82 | 6/9 | 66.7 |
| protein coding | UFM1 | 1/9 | 11.1 | |
| lncRNA | SNHG5 | 1/9 | 11.1 | |
| sense intronic | SNORD91B | 1/9 | 11.1 | |
| down | protein coding | POLR2A, EHMT1, TRAPPC12, ZNF592, YBX1, MAU2, GIT2 | 7/12 | 58.3 |
| misc RNA | RN7SKP208, RN7SKP286, RN7SKP45, RN7SKP7 | 4/12 | 33.3 | |
| lncRNA | AC092620.2 | 1/12 | 8.3 | |
| LEAD vs. CVD | ||||
| up | pseudogene | AC078899.1, AC104651.2, ACTR3P2, ANXA2P2, API5P1, CALM2P2, CALM2P4, CAP1P2, CDC42P6, CFL1P4, CTB-52I2.4, CTNNA1P1, DYNC1I2P1, EIF3FP3, EIF4A1P10, HNRNPA1P7, HSP90B2P, HSP90B3P, HSPA9P1, MSNP1, PDIA3P1, PGDP1, PSME2P2, RP11-1033A18.1, RP11-262D11.2, RP11-286H14.4, RP11-334L9.1, RP11-490H24.5, RP11-6B6.3, RP13-104F24.3, SDCBPP2, SETP14, SRRM1P3 | 33/43 | 76.7 |
| protein coding | ARL6IP1, C1orf216, C1QB, EIF3C, FCGR3B, GLUD2, MNDA, PSME1, S100A10, S100A12 | 10/43 | 23.3 | |
| down | protein coding | D2HGDH, DNAH1, FAM167A, GLI4, HECTD4, HIP1R, PAM16, PIDD1, POLRMT, PPP6R2, RASGRP2, SGSM3, TECPR1, TSC2 | 14/15 | 93.3 |
| pseudogene | TBC1D27P | 1/15 | 6.7 | |
| AAA vs. CVD | ||||
| down | snoRNA | SNORA11, SNORA14B, SNORA60, SNORD127, SNORD64, SNORD94 | 6/10 | 60 |
| protein coding | MALT1, STMN3, TCP11L2 | 3/10 | 30 | |
| miRNA | MIR150 | 1/10 | 10 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Kołodziej, P.; Szymańska, J.; Płachno, B.J.; Zubilewicz, T.; Feldo, M.; et al. Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens. Int. J. Mol. Sci. 2021, 22, 3200. https://doi.org/10.3390/ijms22063200
Zalewski DP, Ruszel KP, Stępniewski A, Gałkowski D, Bogucki J, Kołodziej P, Szymańska J, Płachno BJ, Zubilewicz T, Feldo M, et al. Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens. International Journal of Molecular Sciences. 2021; 22(6):3200. https://doi.org/10.3390/ijms22063200
Chicago/Turabian StyleZalewski, Daniel P., Karol P. Ruszel, Andrzej Stępniewski, Dariusz Gałkowski, Jacek Bogucki, Przemysław Kołodziej, Jolanta Szymańska, Bartosz J. Płachno, Tomasz Zubilewicz, Marcin Feldo, and et al. 2021. "Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens" International Journal of Molecular Sciences 22, no. 6: 3200. https://doi.org/10.3390/ijms22063200
APA StyleZalewski, D. P., Ruszel, K. P., Stępniewski, A., Gałkowski, D., Bogucki, J., Kołodziej, P., Szymańska, J., Płachno, B. J., Zubilewicz, T., Feldo, M., Kocki, J., & Bogucka-Kocka, A. (2021). Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens. International Journal of Molecular Sciences, 22(6), 3200. https://doi.org/10.3390/ijms22063200