
Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
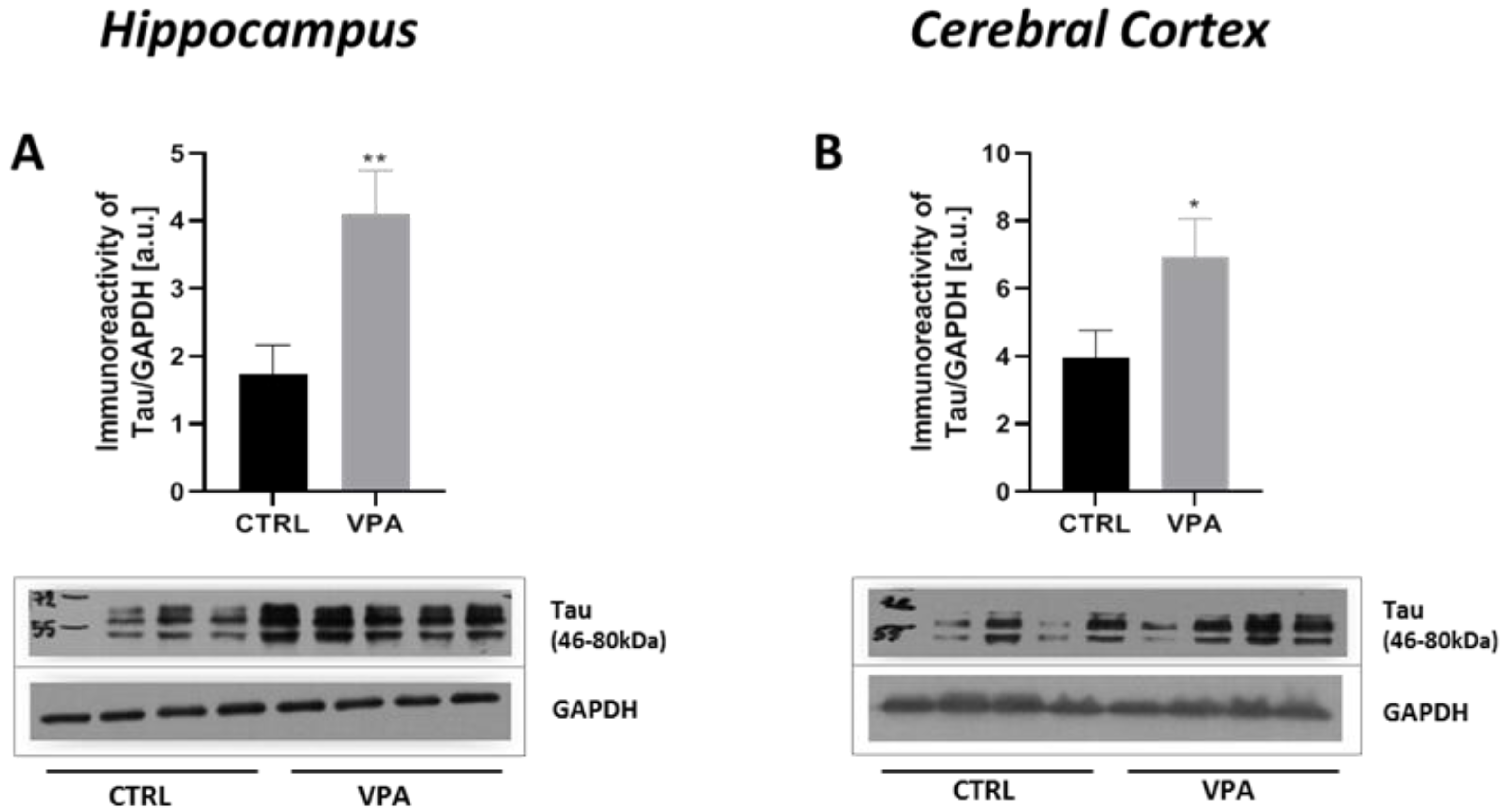
2.1. Prenatal Exposure to VPA Induced Changes in the Level of α/β-Tubulin and MAP-Tau
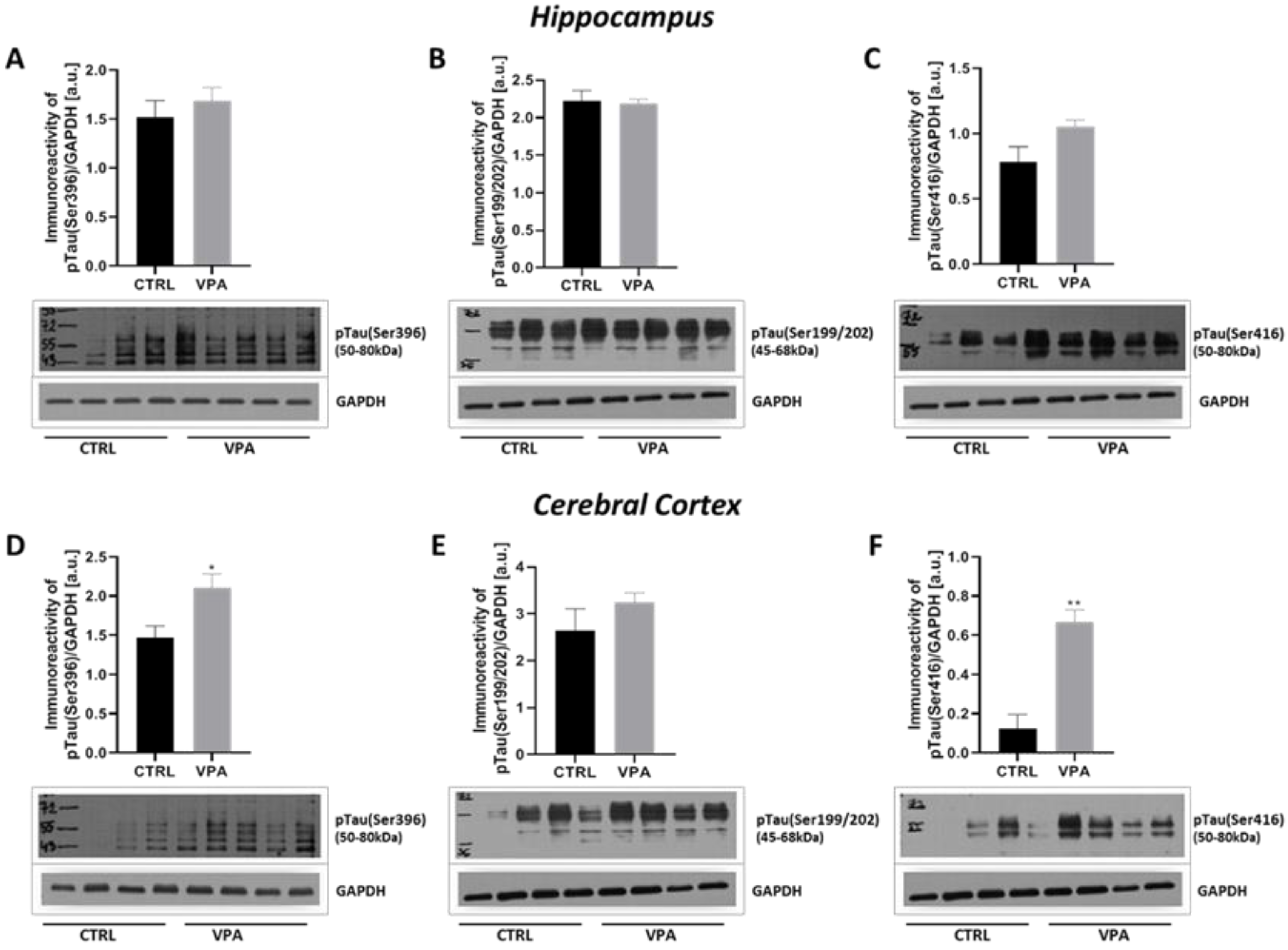
2.2. Prenatal Exposure to VPA Evoked Changes in the Phosphorylation Status of MAP-Tau Protein
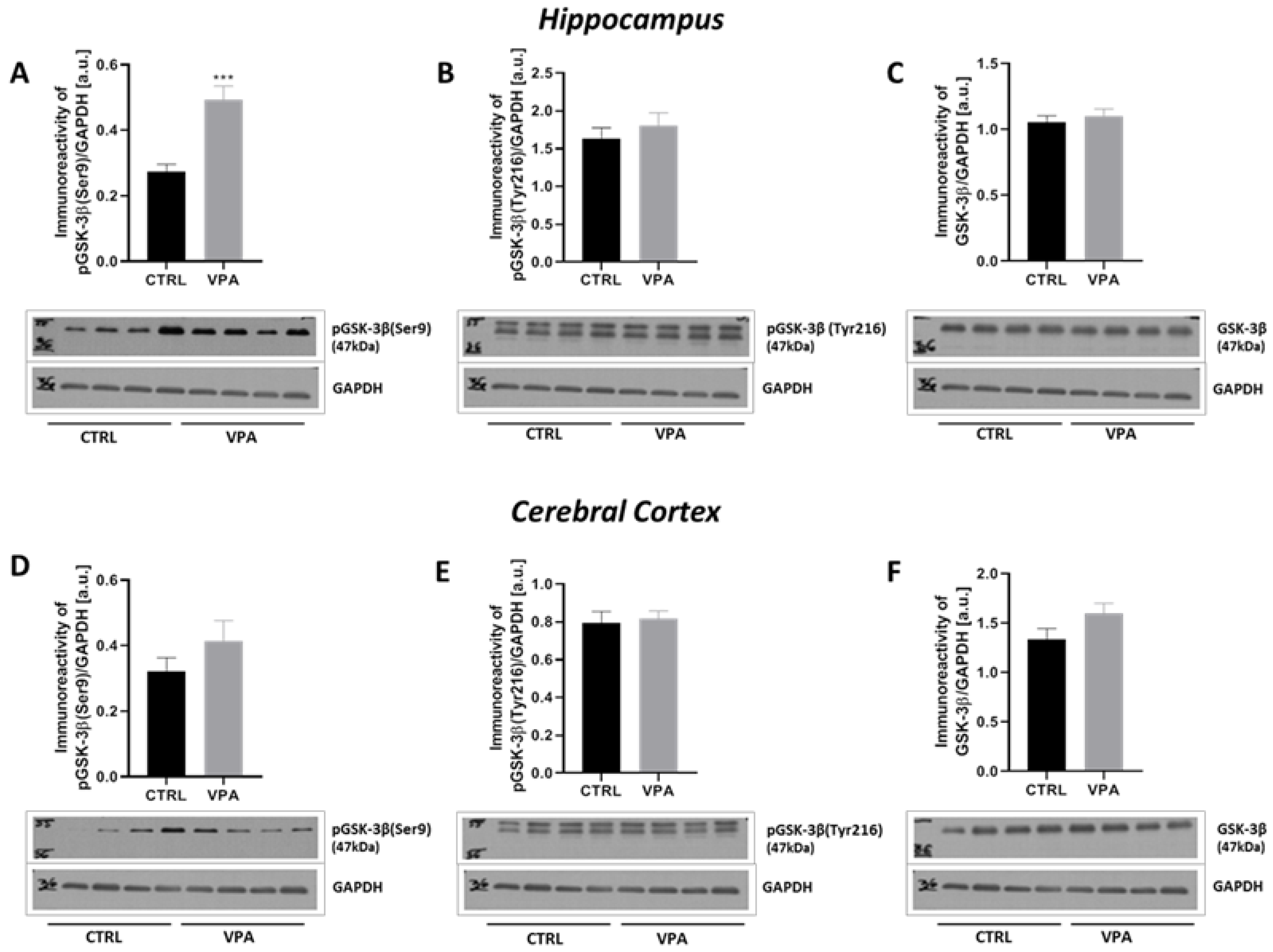
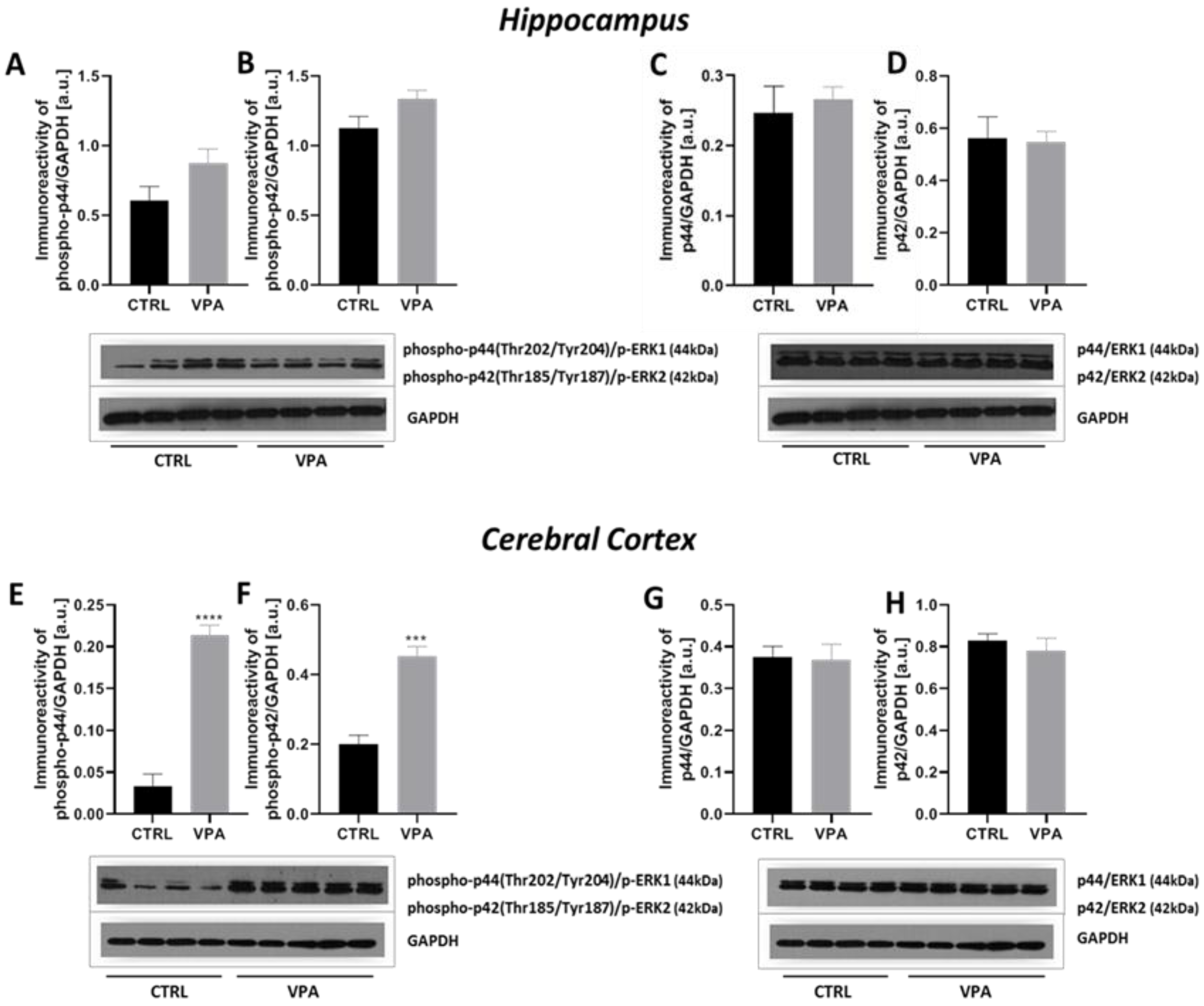
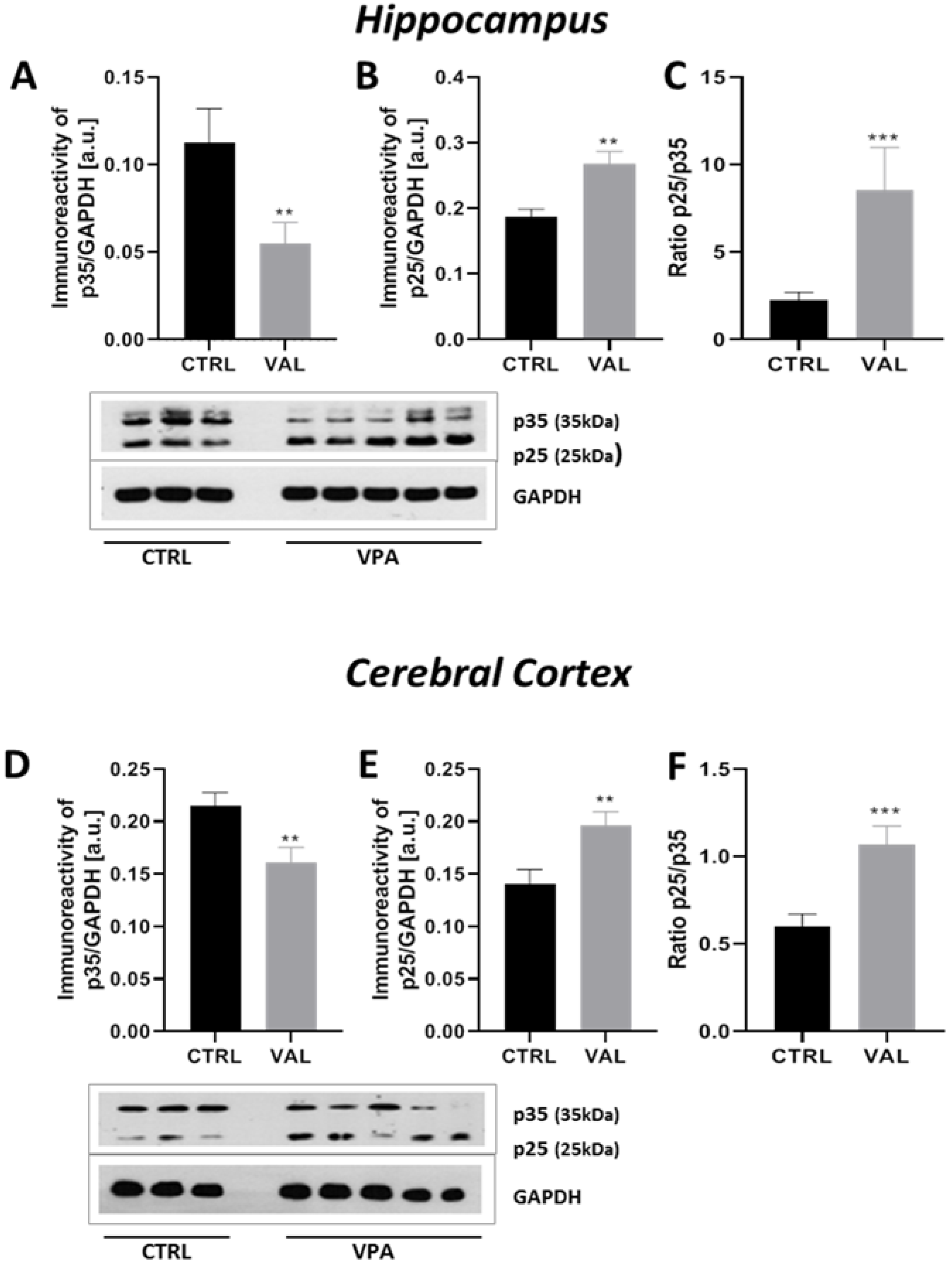
2.3. Prenatal Exposure to VPA Led to the Deregulation of Tau Kinases Activity in a Brain Region-Specific Manner
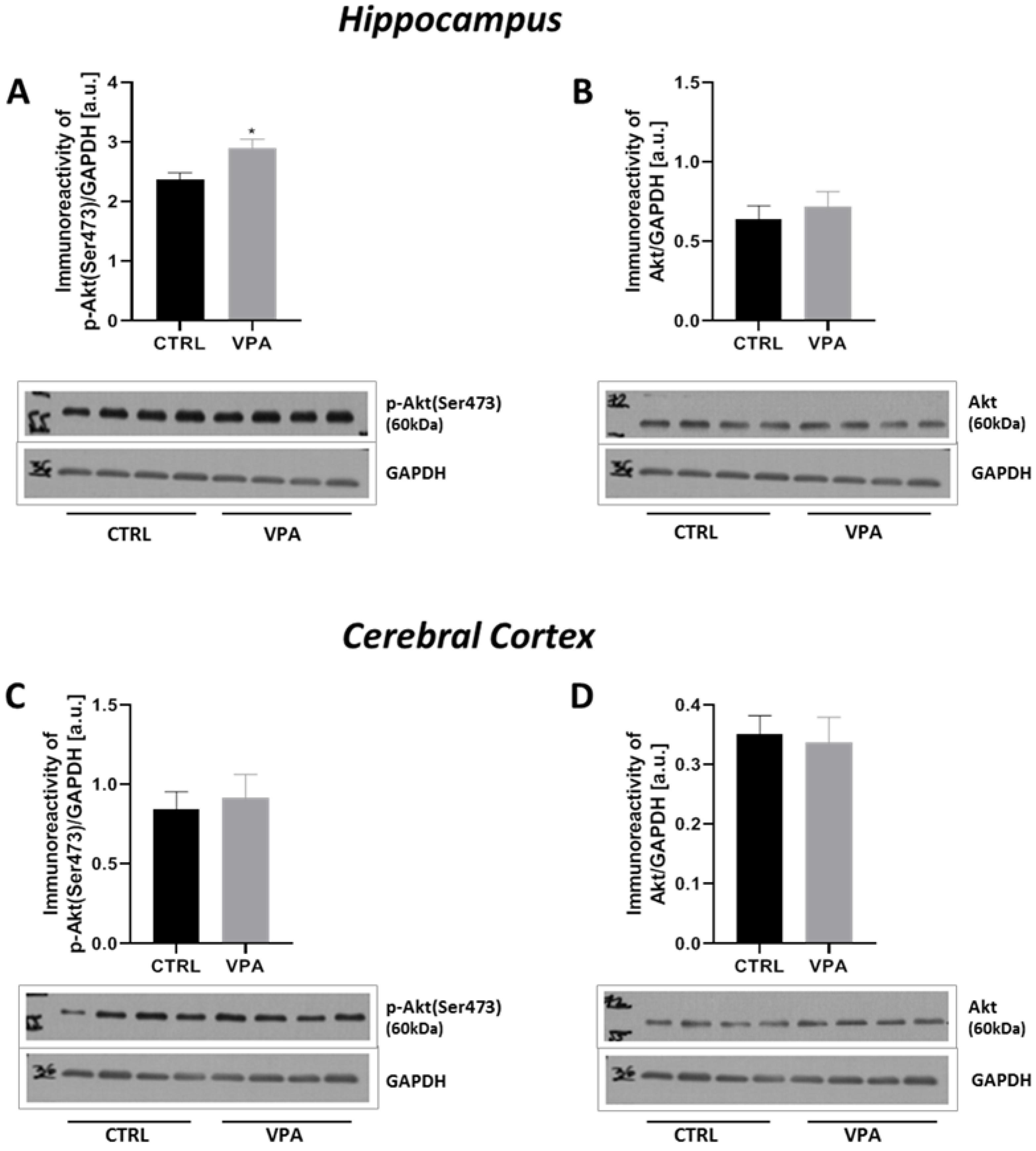
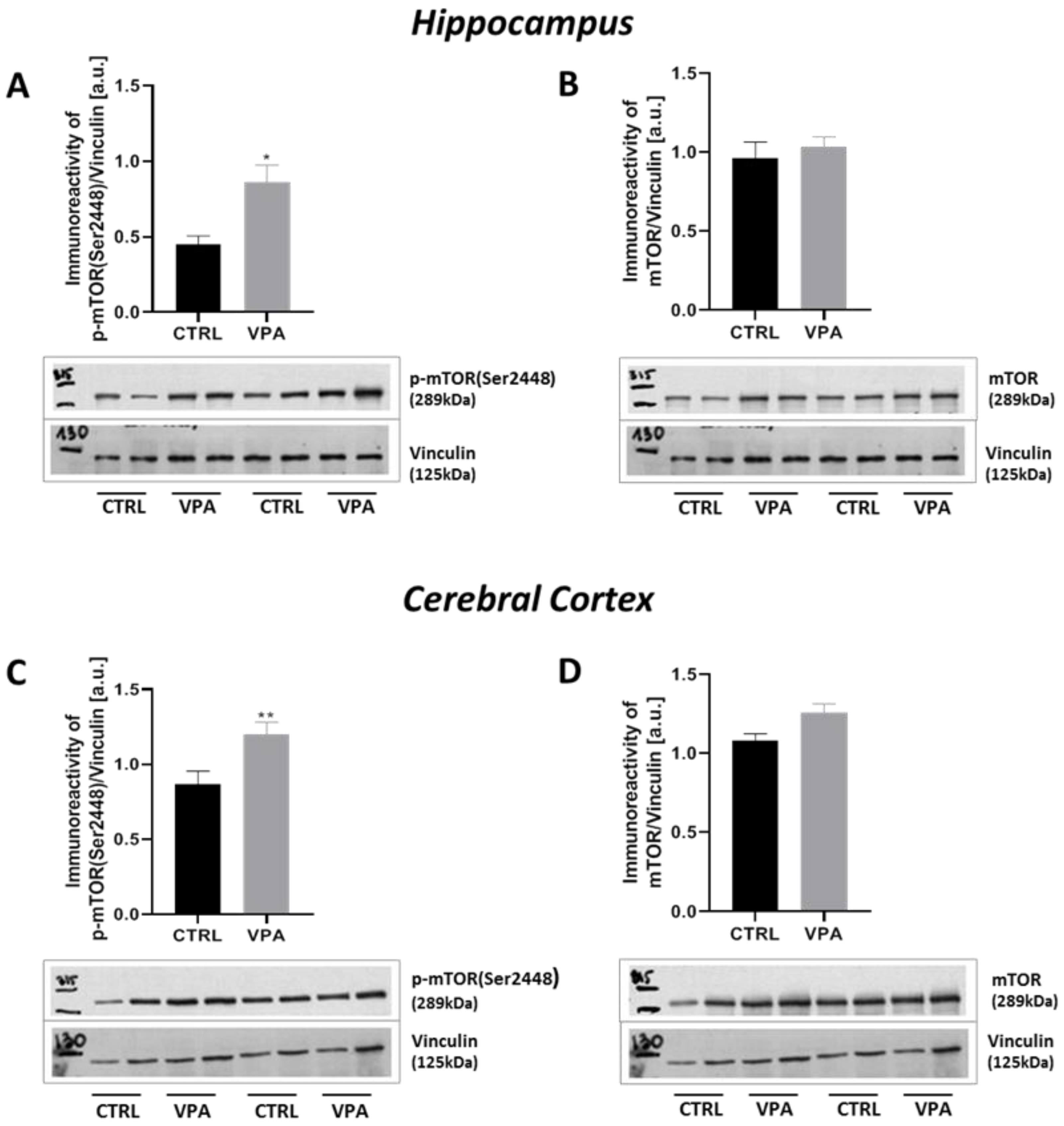
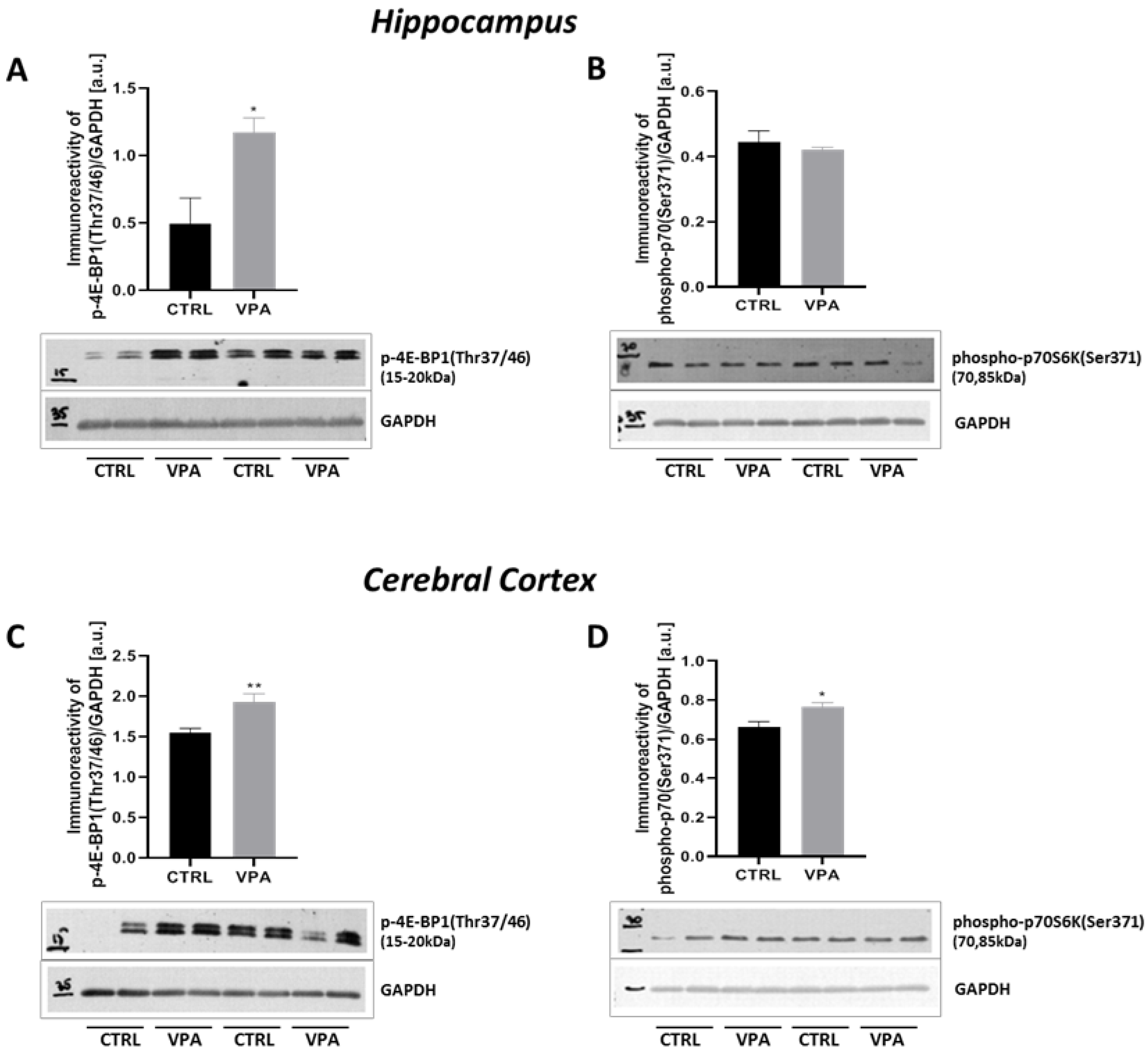
2.4. Prenatal Exposure to VPA Up-Regulated the mTOR Signalling
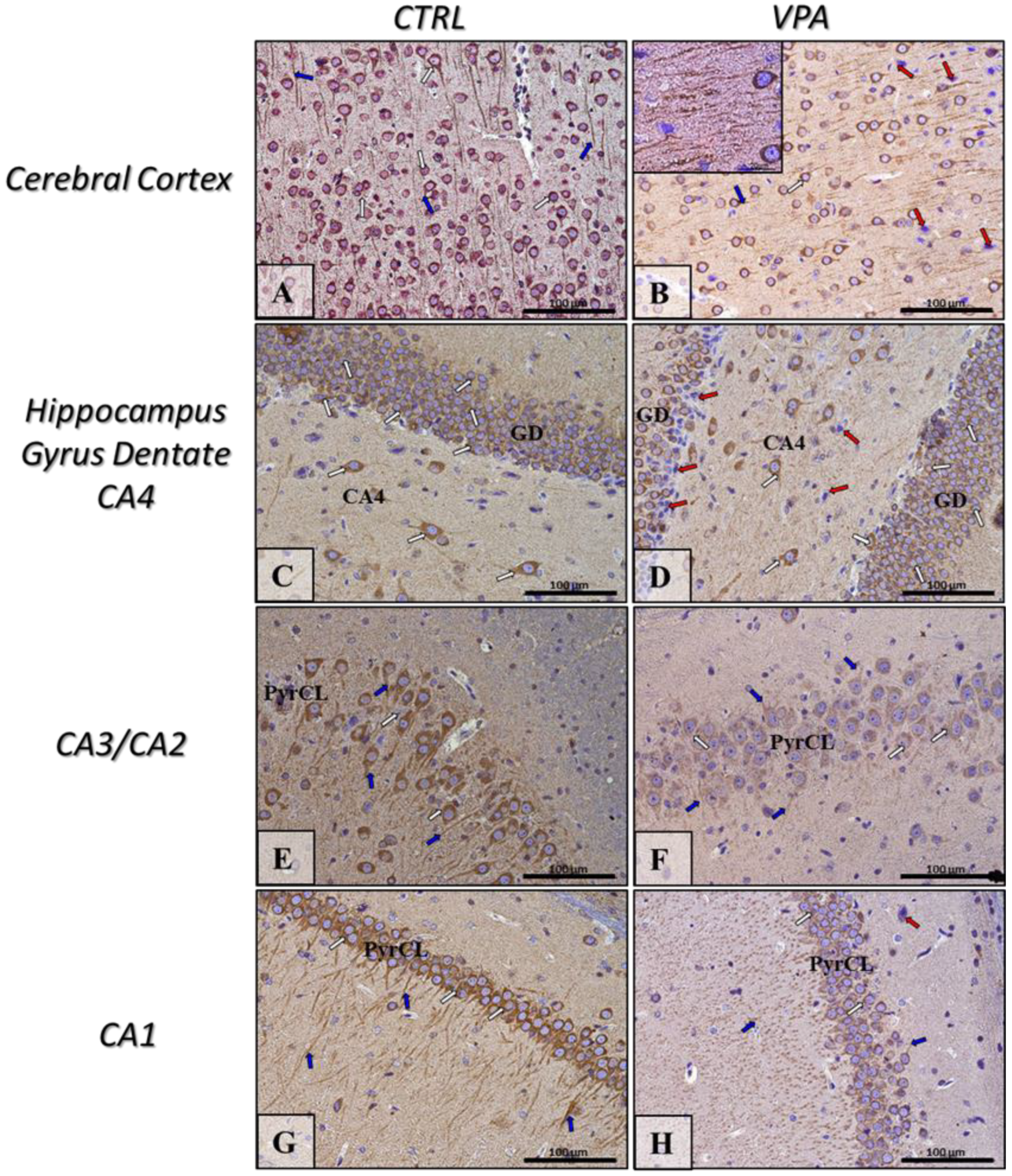
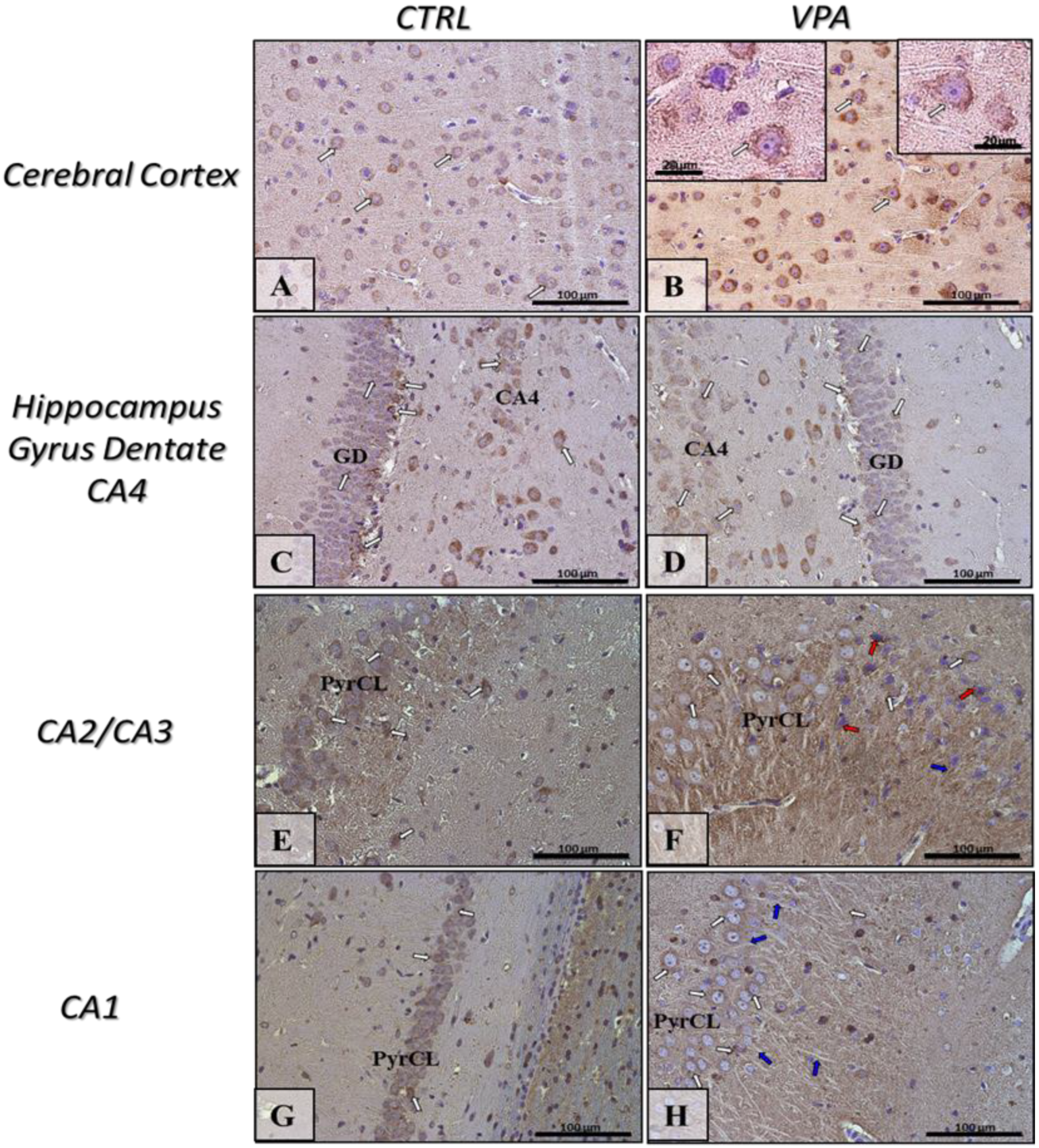
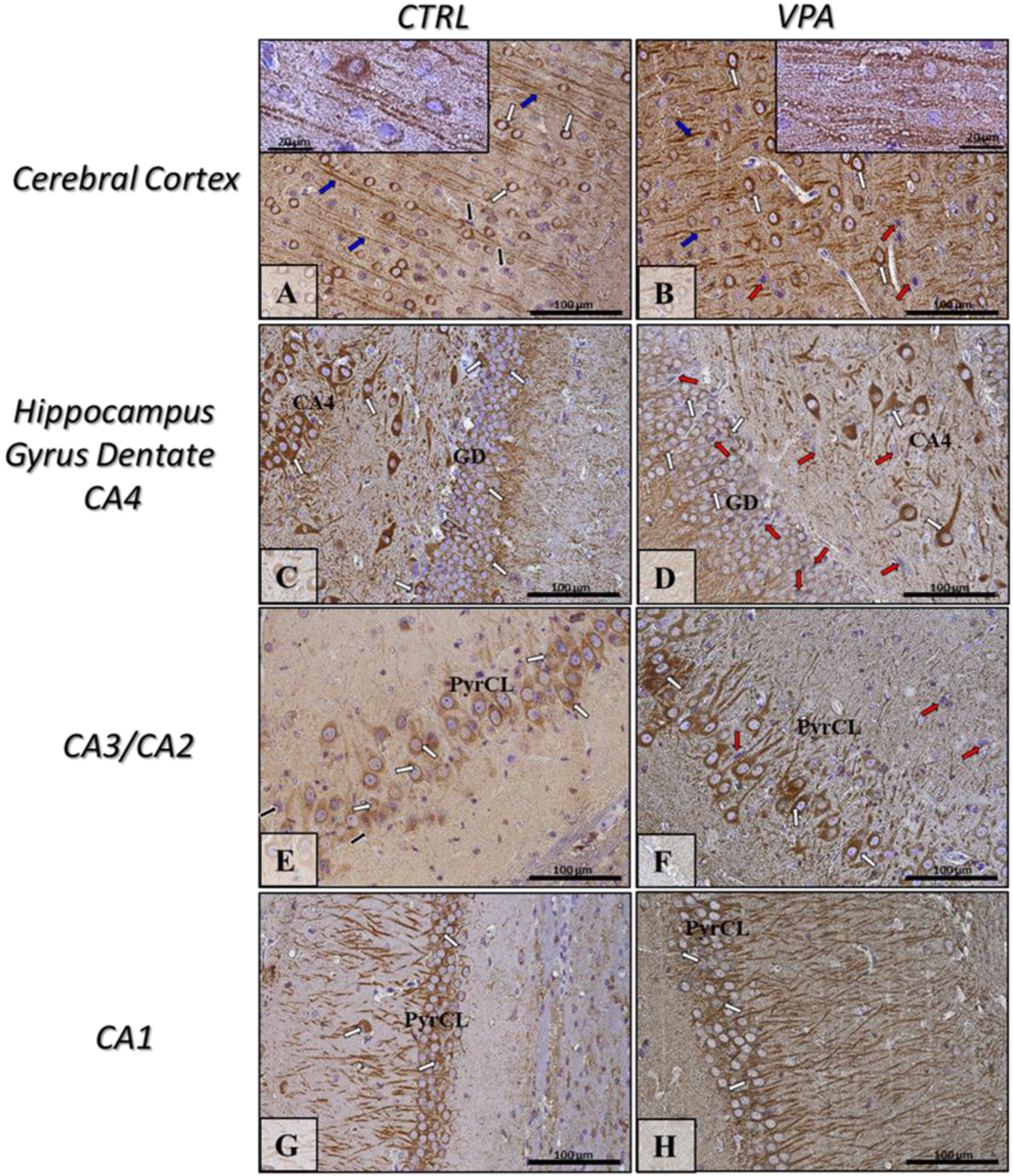
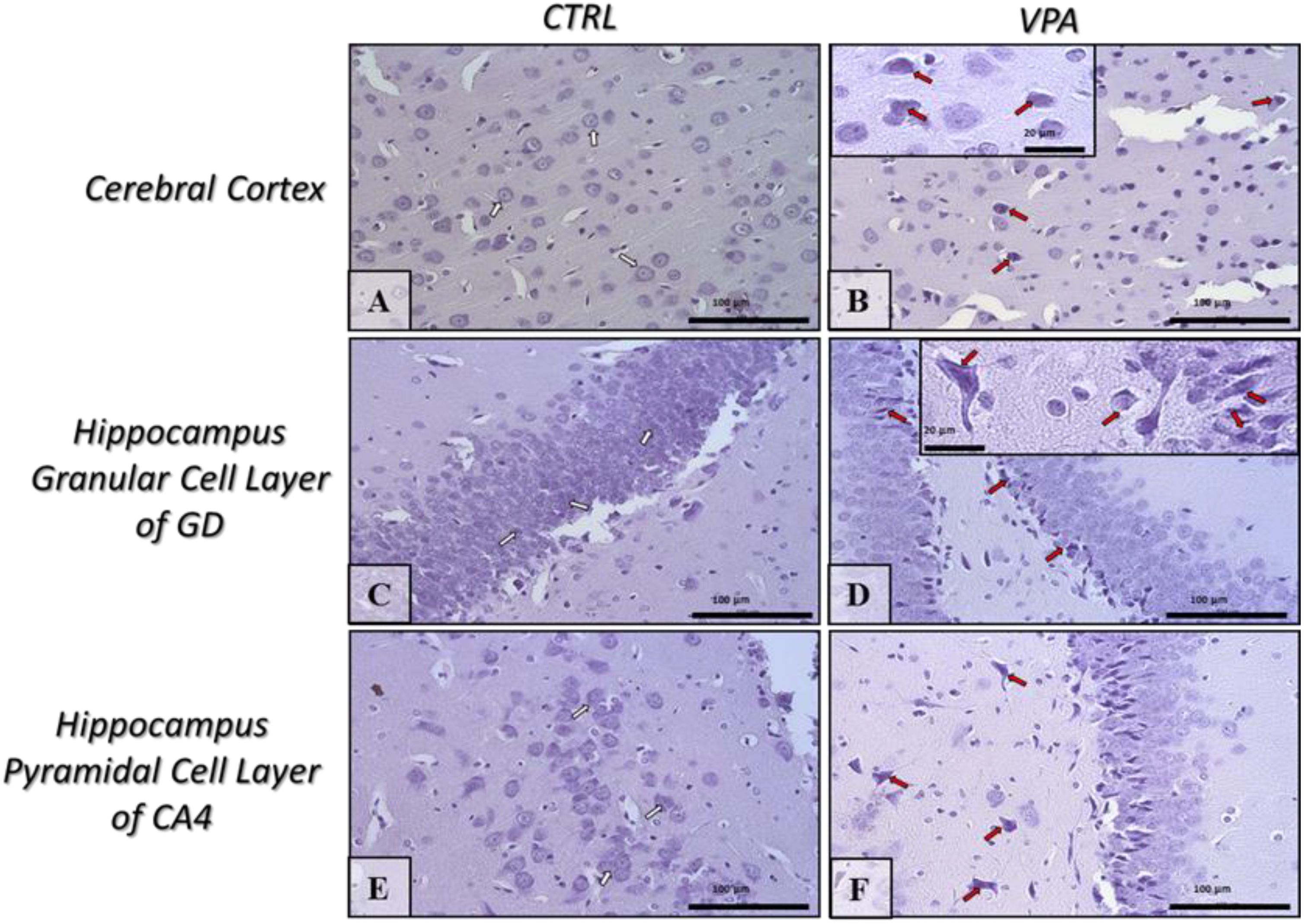
2.5. Prenatal Exposure to VPA Induced Histopathological Changes in the Neurons of Rat Brain
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Animals—In Vivo Model of ASD
4.3. Immunochemical Determination of Protein Levels (Western Blot Analysis)
4.4. Immunohistochemistry Analysis
4.5. Statistical Analysis of Biochemical and Behavioral Results
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism Spectrum Disorder |
| VPA | Valproic acid |
| MTs | Microtubules |
| MAPs | Microtubule-associated proteins |
| Tau | Tubulin-associated unit |
| GSK-3β | Glycogen synthase kinase-3β |
| MAPK | Mitogen-activated protein kinase |
| CDK5 | Cyclin-dependent kinase 5 |
| PKA | cAMP-dependent protein kinase A |
| PKC | Protein kinase C |
| NFTs | Neurofibrillary tangles |
| mTOR | Mechanistic target of rapamycin/mammalian target of rapamycin |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular signal-regulated kinase |
| ERK1 | Extracellular signal-regulated kinase 1 |
| ERK2 | Extracellular signal-regulated kinase 2 |
| SBDP | αII-spectrin breakdown product |
| mTORC1 | mTOR complex 1/Mechanistic target of rapamycin complex 1 |
| p70S6K | p70 ribosomal protein S6 kinase |
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| eIF4E | Eukaryotic translation initiation factor 4E |
| ID | Intellectual disabilities |
| Pb | Lead |
| AD | Alzheimer’s disease |
| PDPKs | Proline-dependent protein kinases |
| LTD | Long-term depression |
| PI3K/Akt | Phosphatidylinositol 3-kinase/protein kinase B |
| RheB1 | GTP-bound levels of small GTPase Ras homolog enriched in brain |
| TSC2 | Tuberous sclerosis complex 2 |
| PTEN | Phosphatase and tensin homolog |
| AGC | Kinase group of kinases related to PKA PKG and PKC |
| PHF | Paired helical filaments |
| PND | Postnatal day |
References
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci. Bull. 2017, 33, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kałużna-Czaplińska, J.; Żurawicz, E.; Jóźwik-Pruska, J. Focus on the social aspect of autism. J. Autism Dev. Disord. 2018, 48, 1861–1867. [Google Scholar] [CrossRef] [Green Version]
- Boorse, J.; Cola, M.; Plate, S.; Yankowitz, L.; Pandey, J.; Schultz, R.T.; Parish-Morris, J. Linguistic markers of autism in girls: Evidence of a “blended phenotyp” during storytelling. Mol. Autism 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, A.E.; Carpentier, P.A.; Babineau, B.A.; Narayan, A.R.; Kielhold, M.L.; Moon, H.M.; Shankar, A.; Su, J.; Saravanapandian, V.; Haditsch, U.; et al. Females are not just ’protected’ males: Sex-specific vulnerabilities in placenta and brain after prenatal immune disruption. eNeuro 2019, 6, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years-Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Bölte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [CrossRef] [Green Version]
- Deckmann, I.; Schwingel, G.B.; Fontes-Dutra, M.; Bambini-Junior, V.; Gottfried, C. Neuroimmune alterationsin autism: A translational analysis focusing on the animal model of autism induced by prenatal exposure to valproic acid. Neuroimmunomodulation 2018, 25, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 2015, 28, 91–102. [Google Scholar] [CrossRef]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology involved in autism spectrum disorder. Front. Cell Neurosci. 2018, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Howell, B.W.; Smith, K.M. Synaptic structural protein dysfunction leads to altered excitation inhibition ratios in models of autism spectrum disorder. Pharmacol. Res. 2019, 139, 207–214. [Google Scholar] [CrossRef]
- Chang, Q.; Yang, H.; Wang, M.; Wei, H.; Hu, F. Role of Microtubule-Associated protein in Autism Spectrum Disorder. Neurosci. Bull. 2018, 34, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Maximo, J.O.; Cadena, E.J.; Kana, R.K. The implications of brain connectivity in the neuropsychology of autism. Neuropsychol. Rev. 2014, 24, 16–31. [Google Scholar] [CrossRef] [Green Version]
- Gąssowska-Dobrowolska, M.; Cieślik, M.; Czapski, G.A.; Jęśko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef]
- Di Martino, A.; Yan, C.G.; Li, Q.; Denio, E.; Castellanos, F.X.; Alaerts, K.; Anderson, J.S.; Assaf, M.; Bookheimer, S.Y.; Daperetto, M.; et al. The autism brain imaging data exchange: Towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol. Psychiatry 2014, 19, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Barón-Mendoza, I.; García, O.; Calvo-Ochoa, E.; Rebollar-García, J.O.; Garzón-Cortés, D.; Haro, R.; González-Arenas, A. Alterations in neuronal cytoskeletal and astrocytic proteins content in the brain of the autistic-like mouse strain C58/J. Neurosci. Lett. 2018, 682, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Trushina, N.I.; Mulkidjanian, A.Y.; Brandt, R. The microtubule skeleton and the evolution of neuronal complexity in vertebrates. Biol. Chem. 2019, 400, 1163–1179. [Google Scholar] [CrossRef]
- Huh, J.W.; Helfaer, M.A.; McIntosh, T.K.; Saatman, K.E. Neurocytoskeletal Changes Following Traumatic Brain Injury. Brain Inj. 2001, 12, 249–265. [Google Scholar]
- Bonini, S.A.; Mastinu, A.; Ferrari-Toninelli, G.; Memo, M. Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. Int. J. Mol. Sci. 2017, 18, 1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Mukaetova-Ladinska, E.B.; Arnold, H.; Jaros, E.; Perry, R.; Perry, E. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol. Appl. Neurobiol. 2004, 30, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and Microtubule Associated Protein Anomalies in Psychiatric Disease. Cytoskeleton 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Wetzel, A.; Grannó, S.; Heaton, G.; Harvey, K. Back to the tubule: Microtubule dynamics in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 409–434. [Google Scholar] [CrossRef] [Green Version]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef] [Green Version]
- Ayaydın, H.; Kirmit, A.; Çelik, H.; Akaltun, I.; Koyuncu, I.; Ulgar, Ş.B. High Serum Levels of Serum 100 Beta Protein, Neuron-specific Enolase, Tau, Active Caspase-3, M30 and M65 in Children with Autism Spectrum Disorders. Clin. Psychopharmacol. Neurosci. 2020, 18, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Jameson, L.; Frey, T.; Zeeberg, B.; Dalldorf, F.; Caplow, M. Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry 1980, 19, 2472–2479. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Li, T.; Paudel, H.K. Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry 2006, 45, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, F.; Suzuki, M.; Shimada, N.; Kagiya, G.; Ohta, E.; Tamura, K.; Maruyama, H.; Ichikawa, T. Stimulatory effect of a-synuclein on the tau-phosphorylation by GSK-3b. FEBS J. 2011, 278, 4895–4904. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Elevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef]
- Muntane, G.; Dalfo, E.; Martinez, A.; Ferrer, I. Phosphorylation of Tau and a-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related a-synucleinopathies. Neuroscience 2008, 152, 913–923. [Google Scholar] [CrossRef]
- Šimić, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Tai, C.; Chang, C.-W.; Yu, G.-Q.; Lopez, I.; Yu, X.; Wang, X.; Guo, W.; Mucke, L. Tau Reduction Prevents Key Features of Autism in Mouse Models. Neuron 2020, 106, 421–437. [Google Scholar] [CrossRef]
- Kadak, M.T.; Cetin, I.; Tarakçıoğlu, M.C.; Özer, Ö.F.; Kaçar, S.; Çimen, B. Low Serum Level α-Synuclein and Tau Protein in Autism Spectrum Disorder Compared to Controls. Neuropediatrics 2015, 46, 410–415. [Google Scholar] [CrossRef]
- Grigg, I.; Ivashko-Pachima, Y.; Hait, T.A.; Korenková, V.; Touloumi, O.; Lagoudaki, R.; Van Dijck, A.; Marusic, Z.; Anicic, M.; Vukovic, J.; et al. Tauopathy in the young autistic brain: Novel biomarker and therapeutic target. Transl. Psychiatry 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Suliman, H.B.; Siniscalco, D.; Antonucci, N.; ElKafrawy, P.; Brahmajothi, M.V. De novo Blood Biomarkers in Autism: Autoantibodies against Neuronal and Glial Proteins. Behav. Sci. 2019, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Modi, M.E.; Sahin, M. Tau: A Novel Entry Point for mTOR-Based Treatments in Autism Spectrum Disorder? Neuron 2020, 106, 359–361. [Google Scholar] [CrossRef]
- Maussion, G.; Carayol, J.; Lepagnol-Bestel, A.M.; Tores, F.; Loe-mie, Y.; Milbreta, U.; Rousseau, F.; Fontaine, K.; Renaud, J.; Moalic, J.-M.; et al. Convergent evidence identifying MAP/microtubule affinity-regulating kinase 1 (MARK1) as a susceptibility gene for autism. Hum. Mol. Genet. 2008, 17, 2541–2551. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Onore, C.; Yang, H.; Van der Water, J.; Ashwood, P. Dynamic Akt/mTOR signalling in children with autism spectrum disorder. Front. Pediatr. 2017, 5, 1–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Alberts, I.; Li, X. Dysregulation of the IGF-I/PI3K/AKT/mTOR signalling pathway in autism spectrum disorders. Int. J. Dev. Neurosci. 2014, 35, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Rosina, E.; Battan, B.; Siracusona, M.; Di Criscio, L.; Hollis, F.; Pacini, L.; Curatolo, P.; Bagni, C. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Sato, A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Bereczki, E.; Zhang, H.; Wang, S.; Li, C.; Ji, X.; Branca, R.M.; Lehtiö, J.; Guan, Z.; Filipcik, P.; et al. Mammalian Target of Rapamycin (mTor) Mediates Tau Protein Dyshomeostasis. J. Biol. Chem. 2013, 288, 15556–15570. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Magrì, A.; Medina, D.X.; Wisely, E.V.; López-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, S.; Natarajan, K.; Curiel, J.; Janke, C.; Liu, J. The emergingrole of the tubulin code: From the tubulin molecule to neuronal function and disease. Cytoskeleton 2016, 73, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Sferra, A.; Nicita, F.; Bertini, E. Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7354. [Google Scholar] [CrossRef] [PubMed]
- Aiken, J.; Buscaglia, G.; Aiken, A.S.; Moore, J.K.; Bates, E.A. Tubulin mutations in brain development disorders: Why haploinsufficiency does not explain TUBA1A tubulinopathies. Cytoskeleton 2020, 77, 40–54. [Google Scholar] [CrossRef]
- Fourel, G.; Boscheron, C. Tubulin mutations in neurodevelopmental disorders as a tool to decipher microtubule function. FEBS Lett. 2020, 594, 3409–3438. [Google Scholar] [CrossRef]
- English, J.A.; Dicker, P.; Focking, M.; Dunn, M.J.; Cotter, D.R. 2-D DIGE analysis implicates cytoskeletal abnormalities in psychiatric disease. Proteomics 2009, 9, 3368–3382. [Google Scholar] [CrossRef]
- Piubelli, C.; Carboni, L.; Becchi, S.; Mathea, A.A.; Domenici, E. Regulation of cytoskeleton machinery, neurogenesis and energy metabolism pathways in a rat gene-environment model of depression revealed by proteomic analysis. Neuroscience 2011, 176, 349–380. [Google Scholar] [CrossRef]
- Summers, B.A.; Cummings, J.F.; DeLahunta, A. Malformations of the central nervous system. In Veterinary Neuropathology; Mobsy: St. Louis, MO, USA, 1995; pp. 68–94. [Google Scholar]
- Moon, L.D.F. Chromatolysis: Do injured axons regenerate poorly when ribonucleases attack rough endoplasmic reticulum, ribosomes and RNA? Dev. Neurobiol. 2018, 78, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhya, T.; Sowjanya, J.; Veeresh, B. Bacopa monniera (L.) Wettst Ameliorates Behavioral Alterations and Oxidative Markers in Sodium Valproate Induced Autism in Rats. Neurochem. Res. 2012, 37, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Maeda, Y.; Kataoka, S.; Ago, Y.; Takuma, K.; Matsuda, T. Effect of prenatal valproic acid exposure on cortical morphology in female mice. J. Pharmacol. Sci. 2012, 118, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Melková, K.; Zapletal, V.; Jansen, S.; Nomilner, E.; Zachrdla, M.; Hritz, J.; Nováˇcek, J.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Functionally specific binding regions of microtubule-associated protein 2cexhibit distinct conformations and dynamics. J. Biol. Chem. 2018, 293, 13297–13309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gąssowska, M.; Czapski, G.A.; Pająk, B.; Cieślik, M.; Lenkiewicz, A.M.; Adamczyk, A. Extracellular α-synuclein leads to microtubule destabilization via GSK-3β-dependent Tau phosphorylation in PC12 cells. PLoS ONE 2014, 9, e94259. [Google Scholar] [CrossRef] [Green Version]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Strużyńska, L.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3β and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 2016, 347, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Mederlyova, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004, 279, 34873–34881. [Google Scholar] [CrossRef] [Green Version]
- Babür, E.; Tan, B.; Delibaş, S.; Yousef, M.; Dursun, N.; Süer, C. Depotentiation of Long-Term Potentiation Is Associated with Epitope-Specific Tau Hyper-/Hypophosphorylation in the Hippocampus of Adult Rats. J. Mol. Neurosci. 2019, 67, 193–203. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.C.; Grundke-Iqbal, I. Cytosolic Abnormally Hyperphosphorylated Tau But Not Paired Helical Filaments Sequester Normal MAPs and Inhibit Microtubule Assembly. J. Alzheimer’s Dis. 2008, 14, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Plana-Ripoll, O.; Pedersen, C.B.; Holtz, Y.; Benros, M.E.; Dalsgaard, S.; de Jonge, P.; Fan, C.C.; Degenhardt, L.; Ganna, A.; Greve, A.N.; et al. Exploring Comorbidity within Mental Disorders among a Danish National Population. JAMA Psychiatry. 2019, 3, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Rhodus, E.K.; Barber, J.; Abner, E.L.; Duff, D.M.C.; Bardach, S.H.; Caban-Holt, A.; Lightner, D.; Rowles, G.D.; Schmitt, F.A.; Jicha, G.A. Behaviors characteristic of Autism spectrum disorder in a geriatric cohort with mild cognitive impairment or early dementia. Alzheimer Dis. Assoc. Disord. 2020, 1, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Rhodus, E.K.; Barber, J.M.; Bardach, S.H.; Nelson, P.T.; Jicha, G.A. Autistic spectrum behaviors in late-life dementia are associated with an increased burden of neurofibrillary tangles in the frontal lobe. Alzheimer’s Dement. 2020, 16, 1–2. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Kaeberlein, M. mTOR Inhibition: From Aging to Autism and Beyond. Scientifica 2013, 2013, 849186. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Dai, X.; Yin, Y. Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol. Cell Neurosci. 2016, 75, 27–35. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.-X.; Zhang, X.-L. PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res. Bull. 2016, 125, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; del C. Alonso, A.; Chen, S.; Omar Chohan, M.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.-J.; Gong, C.-X.; An, W.-L.; Winblad, B.; Cowburn, R.F.; Grundke-Iqbal, I.; Iqba, K. Okadaic-Acid-Induced Inhibition of Protein Phosphatase 2A Produces Activation of Mitogen-Activated Protein Kinases ERK1/2, MEK1/2, and p70 S6, Similar to That in Alzheimer’s Disease. Am. J. Pathol. 2003, 163, 845–858. [Google Scholar] [CrossRef]
- Pei, J.-J.; An, W.-L.; Zhou, X.-W.; Nishimura, T.; Norberg, J.; Benedikz, E.; Götz, J.; Winblad, B. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006, 580, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, S.; Yuan, X.; Hu, Z.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic Acid Promotes Human Glioma U87 Cells Apoptosis and Inhibits Glycogen Synthase Kinase-3β Through ERK/Akt Signaling. Cell. Physiol. Biochem. 2016, 39, 2173–2185. [Google Scholar] [CrossRef]
- De Sarno, P.; Li, X.; Jope, R.S. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 2002, 43, 1158–1164. [Google Scholar] [CrossRef]
- Hu, J.-P.; Xie, J.-W.; Wang, C.-J.; Wang, T.; Wang, X.; Wang, S.-L.; Teng, W.-P.; Wang, Z.-Y. Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res. Bull. 2011, 85, 194–200. [Google Scholar] [CrossRef]
- Mahmood, U.; Ahn, S.; Yang, E.-J.; Choi, M.; Kim, H.; Regan, F.; Kwangwook, C.; Kim, H.-S. Dendritic spine anomalies and PTEN alterations in a mouse model of VPA-induced autism spectrum disorder. Pharmacol. Res. 2018, 128, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Gassowska, M.; Wilkaniec, A.; Chalimoniuk, M.; Strosznajder, J.B.; Adamczyk, A. The mechanisms regulating cyclin-dependentkinase 5 in hippocampus during systemic inflammatory response: The effect on inflammatory gene expression. Neurochem. Int. 2016, 93, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Cai, S.-L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef]
- Engmann, O.; Giese, K.P. Crosstalk between Cdk5 and GSK3 beta: Implications for Alzheimer’s disease. Front. Mol. Neurosci. 2009, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Prabakaran, S.; Cantrelle, F.-X.; Chambraud, B.; Gunawardena, J.; Lippens, G.; Landrieu, I. Characterization of Neuronal Tau Protein as a Target of Extracellular Signal-regulated Kinase. J. Biol. Chem. 2016, 291, 7742–7753. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Sheikh, A.M.; Malik, M.; Wen, G.; Zou, H.; Brown, W.T.; Li, X. Upregulation of Ras/Raf/ERK1/2 signaling and ERK5 in the brain of autistic subjects. Genes Brain Behav. 2011, 10, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Pucilowska, J.; Vithayathil, J.; Pagani, M.; Kelly, C.; Karlo, C.; Robol, C.; Morella, I.; Gozzi, A.; Brambilla, R.; Landreth, G.E. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J. Neurosci. 2018, 38, 6640–6652. [Google Scholar] [CrossRef]
- Russo, A.; Mensah, A.; Bowman, J. Decreased Phosphorylated ERK 1/2 in Individuals with Autism. Int. Ped. Chi. Care 2019, 2, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Borrie, S.C.; Brems, H.; Legius, E.; Bagni, C. Cognitive Dysfunctions in intellectual 671 disabilities: The contributions of the Ras-MAPK and PI3K-AKT-mTOR pathways. Annu. Rev. Genom. Hum. Genet. 2017, 18, 115–142. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Lavillaureix, A.; Yeh, E.; Traglia, M.; Tsang, K.; Bearden, C.E.; Rauen, K.A.; Weiss, L.A. Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. PLoS Genet. 2017, 13, e1006516. [Google Scholar] [CrossRef]
- Wen, Y.; Alshikho, M.J.; Herbert, M.R. Pathway network analyses for autism reveal 823 multisystem involvement, major overlaps with other diseases and convergence upon MAPK 824 and calcium signaling. PLoS ONE 2016, 7, e0153329. [Google Scholar] [CrossRef]
- Fasano, S.; Brambilla, R. Ras-ERK Signaling in Behavior: Old Questions and New Perspectives. Front. Behav. Neurosci. 2011, 5, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Suhan, T.; Michaelis, U.R.; Beek, K.; Rothweiler, F.; Tausch, L.; Werz, O.; Eikel, D.; Zörnig, M.; Nau, H.; et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 2006, 13, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Kim, J.W.; Seung, H.; Kim, K.C.; Gonzales, E.L.T.; Oh, H.A.; Min Yang, S.; Jung Ko, M.; Han, S.-H.; Banerjee, S.; Young Shin, C. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology 2017, 113, 71–81. [Google Scholar] [CrossRef]
- Faridar, A.; Jones-Davis, D.; Rider, E.; Li, J.; Gobius, I.; Morcom, L.; Richards, L.J.; Sen, S.; Sherr, E.H. Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol. Autism 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Shi, H.; Zhao, Y. Phosphorylation of microtubule-associated protein tau by mitogen-activated protein kinase in Alzheimer’s disease. IOP Conf. Ser. Mater. Sci. Eng. 2018, 394, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimer’s Dis. 2013, 33, 123–139. [Google Scholar] [CrossRef]
- Melick, C.H.; Jewell, J.L. Regulation of mTORC1 by Upstream Stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef]
- Parrales, A.; López, E.; Lee-Rivera, I.; López-Colomé, A.M. ERK1/2-dependent activation of mTOR/mTORC1/p70S6K regulates thrombin-induced RPE cell proliferation. Cell. Signal. 2013, 25, 829–838. [Google Scholar] [CrossRef]
- Guégan, J.P.; Ezan, F.; Gailhouste, L.; Langouët, S.; Baffet, G. MEK1/2 Overactivation Can Promote Growth Arrest by Mediating ERK1/2-Dependent Phosphorylation of p70S6K. J. Cell. Physiol. 2014, 229, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, W.-L.; Bjorkdahl, C.; Liu, R.; Cowburn, R.F.; Winblad, B.; Pei, J.-J. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3b in SH-SY5Y neuroblastoma cells. J. Neurochem. 2005, 92, 1104–1115. [Google Scholar] [CrossRef]
- An, W.-L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.-G.; Winblad, B.; Pei, J.-J. Up-Regulation of Phosphorylated/Activated p70 S6 Kinase and Its Relationship to Neurofibrillary Pathology in Alzheimer’s Disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Kim, K.C.; Kim, P.; Go, H.S.; Choi, C.S.; Park, J.H.; Kim, H.J.; Jeon, S.J.; Dela Pena, I.C.; Han, S.H.; Cheong, J.H.; et al. Male-specific alteration in excitatory post-synaptic development and social interaction in pre-natal valproic acid exposure model of autism spectrum disorder. J. Neurochem. 2013, 6, 832–843. [Google Scholar] [CrossRef]
- Mowery, T.M.; Wilson, S.M.; Kostylev, P.V.; Dina, B.; Buchholz, J.B.; Prieto, A.L.; Garraghty, P.E. Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way. Int. J. Dev. Neurosci. 2015, 40, 15–23. [Google Scholar] [CrossRef]
- Kazlauskas, N.; Seiffe, A.; Campolongo, M.; Zappala, C.; Depino, A.M. Sex-specific effects of prenatal valproic acid exposure on sociability and neuroinflammation: Relevance for susceptibility and resilience in autism. Psychoneuroendocrinology 2019, 110, 104441. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Przewłocki, R. Behavioral alterations in rats prenatally to valproic acid: Animal model of autism. Neuropsychopharmacology 2005, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Brand/Cat # | Dilution |
|---|---|---|
| Rabbit anti-α/β-tubulin | Cell Signalling 2148S | 1:1000 5% BSA in TBS-T 0.1% |
| Mouse anti-Tau | Santa Cruz Biotechnology sc-32274 | 1:500 5% milk in TBS-T 0.1% |
| Mouse anti-pTau(Ser396) | Cell Signalling 9632 | 1:250 TBS-T 0.1% |
| Rabbit anti-pTau(Ser199/202) | Sigma T6819 | 1:1000 5% milk in TBS-T 0.1% |
| Rabbit anti-pTau(Ser416) | Cell Signalling 15013P | 1:1000 5% milk in TBS-T 0.1% |
| Mouse anti-pGsk-3β(Ser9) | Santa Cruz Biotechnology sc-373800 | 1:250 5% milk in TBS-T 0.1% |
| Mouse anti-pGsk-3β(Tyr216) | BD Diagnostic 612313 | 1:250 0.1% BSA in TBS-T 0.1% |
| Rabbit anti-Gsk-3β | Cell Signalling 9315 | 1:1000 5% milk in TBS-T 0.1% |
| Rabbit anti-pAkt(Ser473) | Cell Signalling 4060 | 1:500 2% BSA in TBS-T 0.1% |
| Rabbit anti-Akt | Cell Signalling 4691 | 1:750 2% BSA in TBS-T 0.1% |
| Mouse anti-pp44/p42MAPK(Thr202/Tyr204) | Cell Signalling 9106 | 1:1000 TBS-T 0.1% |
| Mouse anti-p44/p42MAPK(Thr202/Tyr204) | Cell Signalling 4696 | 1:1000 5% milk in TBS-T 0.1% |
| Rabbit anti-p35/p25 | Cell Signalling 2680 and Santa Cruz Biotechnology sc-820 (Mix) | 1:1000 1% BSA in TBS-T 0.1% |
| Mouse anti-αII-spectrin | Santa Cruz Biotechnology sc-46696 | 1:1000 5% milk in TBS-T 0.1% |
| Rabbit anti-p-mTOR(Ser2448) | Cell Signalling 5536 | 1:500 TBS-T 0.1% |
| Rabbit anti-mTOR | Cell Signalling 2983 | 1:500 TBS-T 0.1% |
| Rabbit anti-p-4E-BP1(Thr37/46) | Cell Signalling 2855 | 1:250 TBS-T 0.1% |
| Rabbit anti-p-p70S6K(Ser371) | Cell Signalling 9208 | 1:250 TBS-T 0.1% |
| Rabbit anti-GAPDH | Sigma-Aldrich G9545-200UL | 1:50,000 5% milk in TBS-T 0.1% |
| Rabbit anti-vinculin | Cell Signalling 13901 | 1:1000 5% milk in TBS-T 0.1% |
| Secondary antibody | Brand/cat # | Dilution |
| anti-mouse IgG | GE Healthcare VXA931V | 1:4000 5% milk in TBS-T 0.1% |
| anti-rabbit IgG | Sigma-Aldrich A0545-1ML | 1:8000 5% milk in TBS-T 0.1% |
| Changes in Neuronal, Cytoskeletal Proteins | ||
|---|---|---|
| Hippocampus | Cerebral Cortex | |
| α/β-tubulin | ↓ | ↓ |
| MAP-Tau | ↑ | ↑ |
| pTau(Ser396) | - | ↑ |
| pTau(Ser199/202) | - | - |
| pTau(Ser416) | - | ↑ |
| αII-spectrin | ↓ | ↓ |
| Changes in Tau kinases involved in Tau phosphorylation | ||
| p-GSK-3β(Ser9) | ↑ | - |
| p-GSK-3β(Tyr216) | - | - |
| GSK-3β | - | - |
| p35 | ↓ | ↓ |
| p25 | ↑ | ↑ |
| p25/p35 | ↑ | ↑ |
| SBDP/αII-spectrin | ↑ | ↑ |
| p-pERK1(Thr202/Tyr204) | - | ↑ |
| pERK1 | - | - |
| p-pERK2(Thr185/Tyr187) | - | ↑ |
| pERK2 | - | - |
| Changes in Akt/mTOR pathway involved in regulation of Tau homeostasis | ||
| p-Akt(Se473) | ↑ | - |
| Akt | - | - |
| p-mTOR(Ser2448) | ↑ | ↑ |
| mTOR | - | - |
| p-4EBP1(Thr37/46) | ↑ | ↑ |
| p-p70S6K(Ser371) | - | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąssowska-Dobrowolska, M.; Kolasa-Wołosiuk, A.; Cieślik, M.; Dominiak, A.; Friedland, K.; Adamczyk, A. Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid. Int. J. Mol. Sci. 2021, 22, 3209. https://doi.org/10.3390/ijms22063209
Gąssowska-Dobrowolska M, Kolasa-Wołosiuk A, Cieślik M, Dominiak A, Friedland K, Adamczyk A. Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid. International Journal of Molecular Sciences. 2021; 22(6):3209. https://doi.org/10.3390/ijms22063209
Chicago/Turabian StyleGąssowska-Dobrowolska, Magdalena, Agnieszka Kolasa-Wołosiuk, Magdalena Cieślik, Agnieszka Dominiak, Kristina Friedland, and Agata Adamczyk. 2021. "Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid" International Journal of Molecular Sciences 22, no. 6: 3209. https://doi.org/10.3390/ijms22063209
APA StyleGąssowska-Dobrowolska, M., Kolasa-Wołosiuk, A., Cieślik, M., Dominiak, A., Friedland, K., & Adamczyk, A. (2021). Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid. International Journal of Molecular Sciences, 22(6), 3209. https://doi.org/10.3390/ijms22063209