
Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19

 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
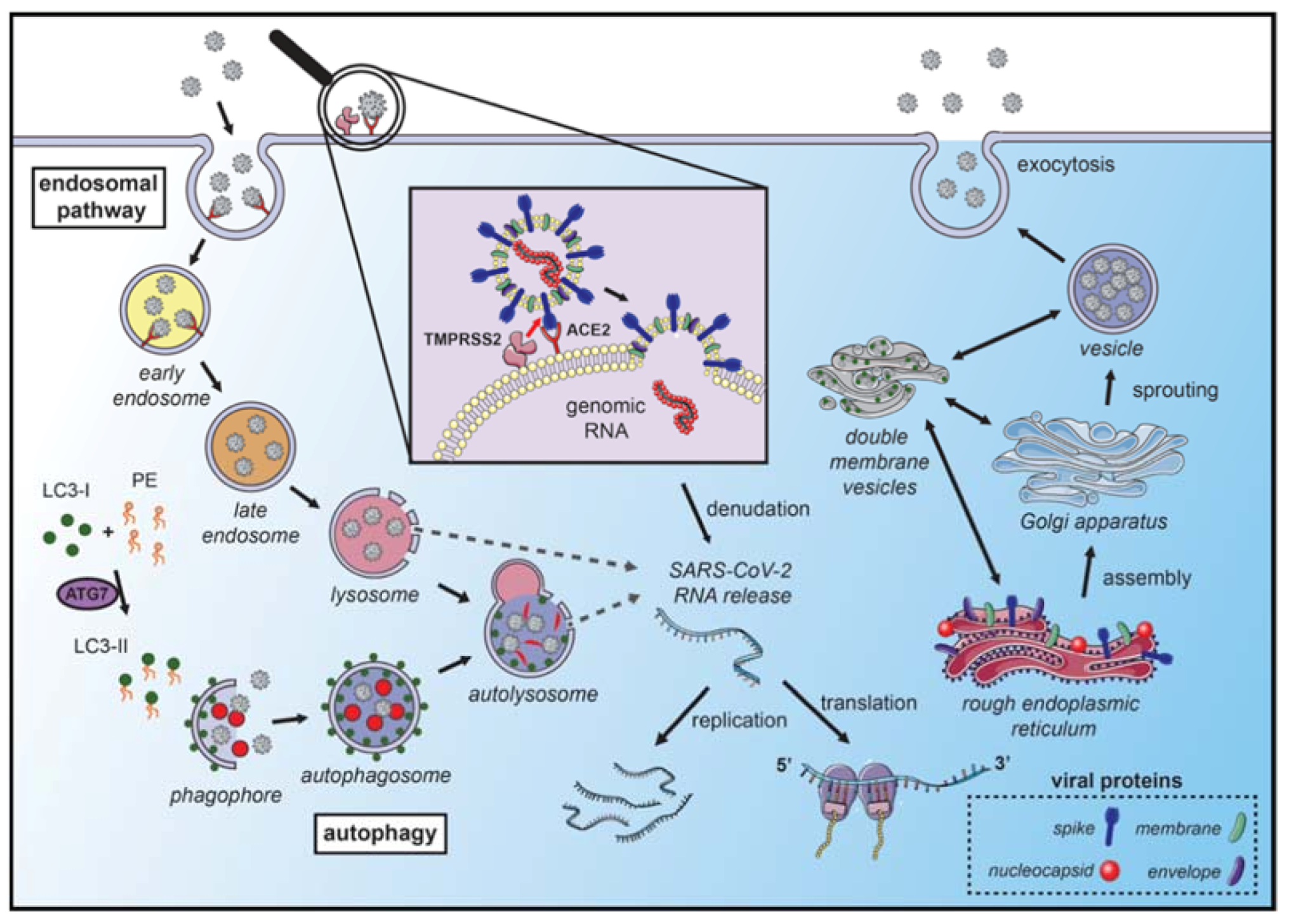
2. Coronavirus: Concepts; Types; Compositions; Mechanisms of Infection and Replication
3. Coronavirus Hijack the Autophagy Machinery to Foster Replication
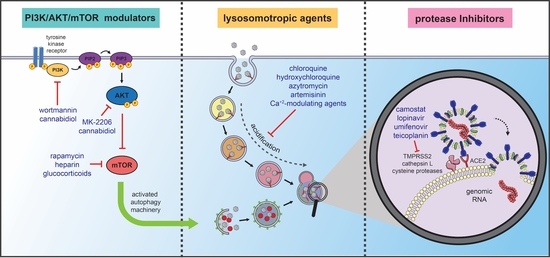
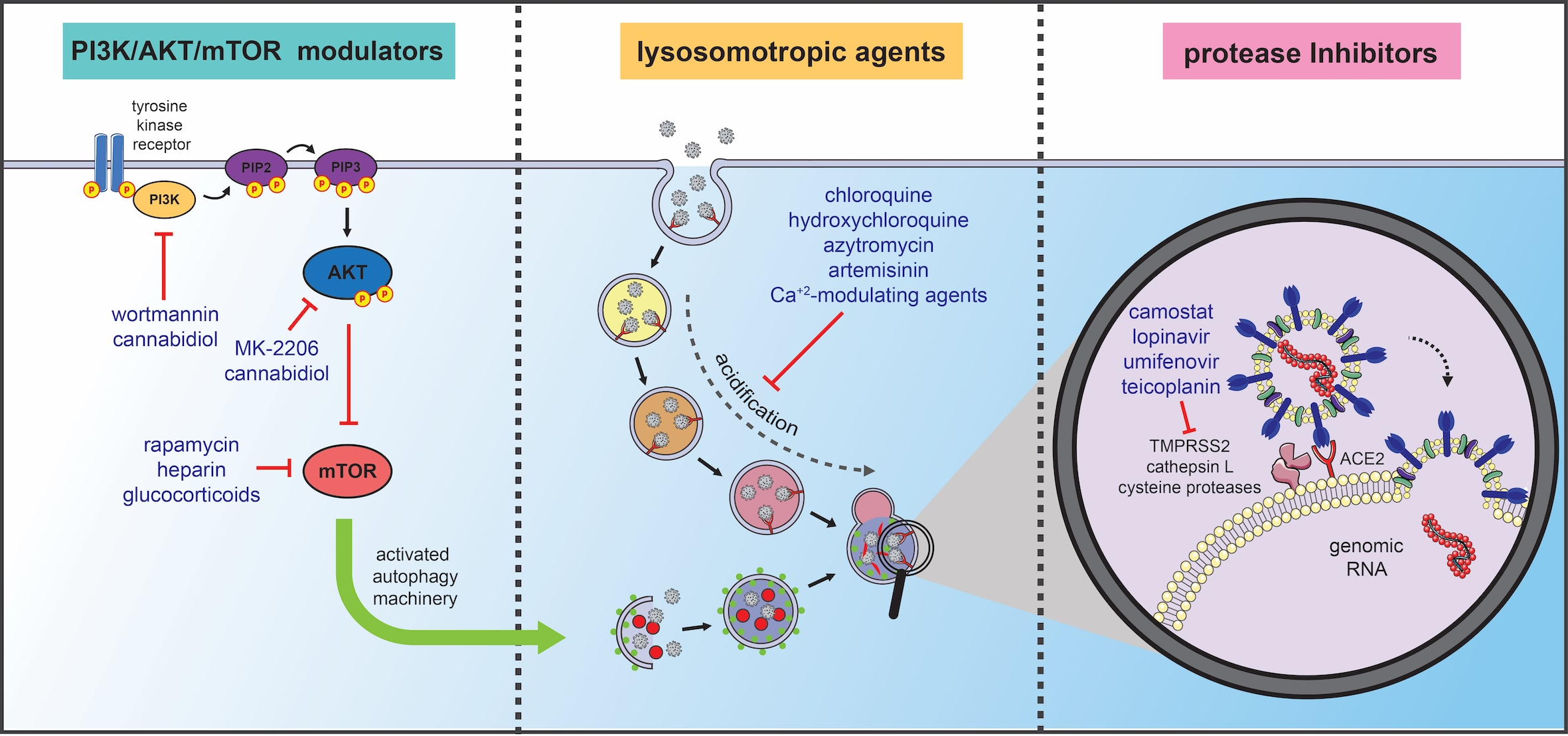
4. Autophagy-Related Therapeutic Targets for COVID-19 Management
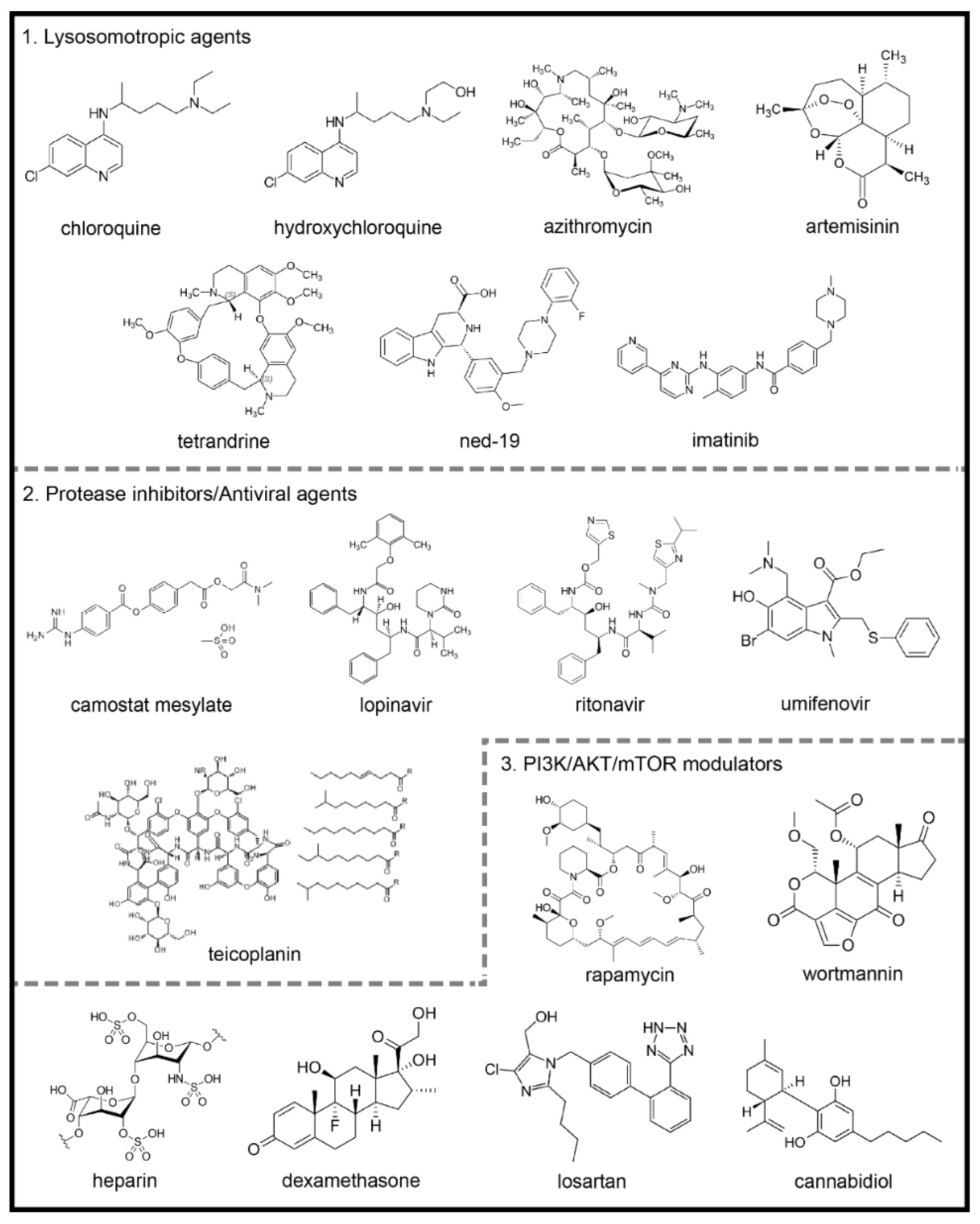
4.1. Lysosomotropic Agents
4.1.1. Chloroquine and Hydroxychloroquine
4.1.2. Azithromycin
4.1.3. Artemisinin and Its Derivative Compounds
4.1.4. Two-Pore Channels Modulating Agents
4.1.5. Imatinib
4.2. Protease Inhibitors/Antiviral Agents: The Prevention of Infection
4.2.1. Camostat Mesylate
4.2.2. Lopinavir
4.2.3. Umifenovir
4.2.4. Teicoplanin and Others
4.3. PI3K/AKT/mTOR Modulators
4.3.1. Rapamycin
4.3.2. Heparin
4.3.3. Glucocorticoids
4.3.4. Angiotensin-Converting Enzyme Inhibitors (IECAs) and Type 1 Angiotensin II Receptors Blockers (ARB)
4.3.5. Cannabidiol
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 2019-nCoV | 2019 novel coronavirus |
| ACE2 | angiotensin-converting enzyme 2 |
| ACEI | angiotensin-converting enzyme inhibitor |
| AKT | serine-threonine kinase |
| Ambra1 | activating molecule in Beclin 1-regulated autophagy |
| AMPK | protein kinase activated by AMP |
| ARB | angiotensin receptor blocker |
| AT1 | type 1 angiotensin II receptor |
| AT2 | type 2 angiotensin II receptor |
| Atg/ATG | autophagy-related protein/gene |
| BCL-2 | B-cell lymphoma 2 |
| BCR-ABL | breakpoint cluster region gene-Abelson proto-oncogene |
| BECN1 | beclin-1 gene |
| CB1 | cannabinoid receptor type 1 |
| Beclin-1/Vps34 | Beclin-1/vacuolar protein sorting 34 |
| CCL | chemokine (C-C motif) ligand |
| COVID-19 | Coronavirus Disease 2019 |
| CoVs | coronavirus |
| CQ | chloroquine |
| DMVs | double-membrane vesicles |
| EBOV | Ebola virus |
| EIF4B | eukaryotic translation initiation factor 4B |
| EIF4F | eukaryotic translation initiation factor 4F |
| EIF4G1 | Eukaryotic Translation Initiation Factor 4 Gamma 1 |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| GABARAP | gamma-aminobutyric acid receptor-associated protein |
| GDI2 | Guanosine nucleotide diphosphate dissociation inhibitor 2 |
| GCs | glucocorticoids |
| GFP | green fluorescent protein |
| H1N1 | influenza A |
| HBV | hepatitis B virus |
| HCMV | human cytomegalovirus |
| HCQ | hydroxychloroquine |
| HCV | hepatitis C virus |
| HIV | human immunodeficiency virus |
| HNE | human nasal epithelial cells |
| HPV | human papilloma virus |
| HRV | human rhinovirus |
| HSV-1/HSV-2 | human herpes simplex virus 1 and 2 |
| IBV | infectious Bronchitis Virus |
| IECAs | angiotensin-converting enzyme inhibitors |
| IL-1β | interleukin-1 β |
| LASV | Lassa virus |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| LIR | LC3-interacting region |
| MAP1LC3 | Microtubule-Associated Protein 1A/1B-Light Chain 3 |
| MAPK | mitogen-activated protein kinase |
| MERS-CoV | Middle-East respiratory syndrome coronavirus |
| MHRA | Medicines and Healthcare products Regulatory Agency |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NAADP | Nicotinic Acid Adenine Dinucleotide Phosphate |
| NF-κB | nuclear factor kappa B |
| NHS | National Health Service |
| NIH | National Institute of Health |
| Nsp | nonstructural protein |
| Orf | open reading frame |
| PEDV | porcine epidemic diarrhea virus |
| PI3K | phosphoinositide 3-kinase |
| PI3P | phosphatidylinositol 3-phosphate |
| PLP2-TM | papain-like protease PLP2 |
| PPARγ | peroxisome proliferator-activated receptor γ |
| RAS | renin-angiotensin system |
| RAB1A | Ras-related protein Rab-1A |
| RAB6A | Ras-related protein Rab-6A |
| RAB6D | Ras-related protein Rab-6D |
| RAB7A | Ras-related protein Rab-7A |
| RECOVERY | Randomised Evaluation of COVID-19 Therapy |
| RNA | ribonucleic acid |
| S | Spike |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SCFD1 | sec1 family domain containing 1 |
| SQSTM1 | sequestosome-1 |
| SKP2 | S-phase kinase-associated protein 2 |
| SNAP29 | synaptosome associated protein 29 |
| SNARE | N-ethylmaleimide-sensitive factor attachment protein receptor |
| STAT3 | signal transducer and activator of transcription 3 |
| Stx17 | Syntaxin 17 |
| TFEB | Transcription Factor EB |
| TGEV | transmissible gastroenteritis virus |
| TMEV | Theiler’s murine encephalomyelitis virus |
| TMPRSS2 | transmembrane serine protease 2 |
| TNF-α | tumor necrosis factor α |
| TPCs | Two-Pore Channels |
| TRIM32 | tripartite motif-containing 32 |
| TRPML1 | transient receptor potential mucolipin 1 |
| ULK1 | Unc-51-like autophagy activating kinase 1 |
| UNC-5 | Netrin receptor UNC-5 (Uncoordinated protein 5) |
| USO1 | General vesicular transport factor p115 |
| USA | United States of America |
| VAMP8 | lysosomal vesicle-associated membrane protein 8 |
| VPS 34 | vacuolar protein sorting 34 |
| VPS 34-IN1 | vacuolar protein sorting 34 inhibitor 1 |
| WHO | World Health Organization |
| WIPI | WD repeat domain phosphoinositide-interacting protein |
| β-CoVs | Betacoronavirus genus |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Kuo, R.L.; Shih, S.R. COVID-19: The first documented coronavirus pandemic in history. Biomed. J. 2020, 43, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Neilson, S. Aylin Woodward Coronavirus: A 1-Year Timeline of the Pandemic Since China’s 1st Case—Business Insider. Available online: https://www.businessinsider.com/coronavirus-pandemic-timeline-history-major-events-2020-3 (accessed on 24 December 2020).
- Sarkar, R.; Mitra, S.; Chandra, P.; Saha, P.; Banerjee, A.; Dutta, S.; Chawla-Sarkar, M. Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: An endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations. Arch. Virol. 2021. [Google Scholar] [CrossRef]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA J. Am. Med. Assoc. 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Liu, K.; Fang, Y.-Y.; Deng, Y.; Liu, W.; Wang, M.-F.; Ma, J.-P.; Xiao, W.; Wang, Y.-N.; Zhong, M.-H.; Li, C.-H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. 2020, 133, 1025–1031. [Google Scholar] [CrossRef]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 105, pp. 93–116. ISBN 9780128184561. [Google Scholar]
- Zhang, N.; Jiang, S.; Du, L. Current advancements and potential strategies in the development of MERS-CoV vaccines. Expert Rev. Vaccines 2014, 13, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, X.; Sun, Y.; Mao, X.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Ding, C.; Liao, Y. Infectious bronchitis virus entry mainly depends on clathrin mediated endocytosis and requires classical endosomal/lysosomal system. Virology 2019, 528, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.M.; Reggiori, F.; De Haan, C.A.M. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458–459, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Oudshoorn, D.; Rijs, K.; Limpens, R.W.A.L.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio 2017, 8, 1658–1675. [Google Scholar] [CrossRef] [Green Version]
- De Haan, C.A.M.; Rottier, P.J.M. Molecular Interactions in the Assembly of Coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar]
- Westerbeck, J.W.; Machamer, C.E. A Coronavirus E Protein Is Present in Two Distinct Pools with Different Effects on Assembly and the Secretory Pathway. J. Virol. 2015, 89, 9313–9323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 210–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Mechanisms of disease: Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. MTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013, 32, 3311–3318. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.J.; De Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, M.; Nishimura, T.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy 2017, 13, 1452–1464. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.T.; Giddings, T.H.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of Cellular Autophagosomal Machinery by RNA Viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, J.; Thomas, S.M. Autophagy-dependent secretion: Mechanism, factors secreted, and disease implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.A.K.; Sany, M.R.U.; Islam, M.S.; Islam, A.B.M.M.K. Epigenetic Regulator miRNA Pattern Differences Among SARS-CoV, SARS-CoV-2, and SARS-CoV-2 World-Wide Isolates Delineated the Mystery Behind the Epic Pathogenicity and Distinct Clinical Characteristics of Pandemic COVID-19. Front. Genet. 2020, 11, 765. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Nabar, N.R.; Shi, C.S.; Kamenyeva, O.; Xiao, X.; Hwang, I.Y.; Wang, M.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Ma, M.; Zheng, Y.; Pan, Y.; Lin, X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 12562–12568. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; MacPherson, S.; Sumpter, R.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy Protects against Sindbis Virus Infection of the Central Nervous System. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Ojha, C.R.; Rodriguez, M.; Karuppan, M.K.M.; Lapierre, J.; Kashanchi, F.; El-Hage, N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PLoS ONE 2019, 14, e0208543. [Google Scholar] [CrossRef]
- Karuppan, M.K.M.; Ojha, C.R.; Rodriguez, M.; Lapierre, J.; Aman, M.J.; Kashanchi, F.; Toborek, M.; Nair, M.; El-Hage, N. Reduced-Beclin1-Expressing Mice Infected with Zika-R103451 and Viral-Associated Pathology during Pregnancy. Viruses 2020, 12, 608. [Google Scholar] [CrossRef]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human papilloma virus and autophagy. Int. J. Mol. Sci. 2018, 19, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the Phosphatidylinositol 3-Kinase/Akt/mTOR Pathway and Inhibition of Autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Ju, M.; Bu, W.; Chen, K.; Li, L.; Li, M.; Chen, X.; Gu, H. HPV Infection Downregulates the Expression of Autophagy-Related Genes in Condylomata Acuminata. Dermatology 2019, 235, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xiong, Y.; Zhang, J.; Shao, T.; Chen, S.; Miao, B.; Wang, Z.; Du, Q.; Huang, Y.; Tong, D. Autophagy promotes porcine parvovirus replication and induces non-apoptotic cell death in porcine placental trophoblasts. Viruses 2019, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.Y.; Huang, H.I. Autophagy is induced and supports virus replication in Enterovirus A71-infected human primary neuronal cells. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Yang, Y.; Li, C.; Chen, J.; Li, Z.; Li, X.; Li, S.; Wu, F.; Hu, Z.; Yang, Z. The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Chiramel, A.; Brady, N.; Bartenschlager, R. Divergent Roles of Autophagy in Virus Infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Dethloff, F.; Emanuel, J.; Weckmann, K.; Heinz, D.E.; Heinemann, N.; Lennarz, M.; Richter, A.; et al. Analysis of SARS-CoV-2-controlled autophagy reveals spermidine, MK-2206, and niclosamide as putative antiviral therapeutics. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Klionsky, D.J. Autophagy/virophagy: A “disposal strategy” to combat COVID-19. Autophagy 2020, 2271–2272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin IV, H.W. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; De Haan, C.A.M.; Molinari, M. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe Acute Respiratory Syndrome Coronavirus Replication Is Severely Impaired by MG132 due to Proteasome-Independent Inhibition of M-Calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Shen, H.M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Holding, A.N.; Giorgi, F.M. Web tools to fight pandemics: The COVID-19 experience. Brief. Bioinform. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Bauer, D.C.; Tay, A.P.; Wilson, L.O.W.; Reti, D.; Hosking, C.; McAuley, A.J.; Pharo, E.; Todd, S.; Stevens, V.; Neave, M.J.; et al. Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS-CoV-2 outbreak. Transbound. Emerg. Dis. 2020, 67, 1453–1462. [Google Scholar] [CrossRef]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Reply to Sánchez-Pacheco et al., Chookajorn, and Mavian et al.: Explaining phylogenetic network analysis of SARSCoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 12524–12525. [Google Scholar] [CrossRef]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Tsai, S.J.; Ai, Y.; Li, M.; Pekosz, A.; Cox, A.; Atai, N.; Gould, S.J. The D614G Mutation Enhances the Lysosomal Trafficking of SARS-CoV-2 Spike. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kuehn, B.M. Genetic Analysis Tracks SARS-CoV-2 Mutations in Human Hosts. JAMA J. Am. Med. Assoc. 2020, 323, 2363. [Google Scholar] [CrossRef] [PubMed]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol. 2020, 1–15. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kliche, J.; Kuss, H.; Ali, M.; Ivarsson, Y. Cytoplasmic short linear motifs in ACE2 and integrin β 3 link SARS-CoV-2 host cell receptors to mediators of endocytosis and autophagy. Sci. Signal. 2021, 14, eabf1117. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.M.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and Origin of Membrane Vesicles Associated with the Severe Acute Respiratory Syndrome Coronavirus Replication Complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy 2020, 1–9. [Google Scholar] [CrossRef]
- Servier Medical Art by Servier Is Licensed under a Creative Commons Attribution 3.0 Unported License. Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 2 January 2021).
- Cong, Y.; Verlhac, P.; Reggiori, F. The Interaction between Nidovirales and Autophagy Components. Viruses 2017, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.D.; Galle-Treger, L.; Akbari, O. Role of Autophagy in Lung Inflammation. Front. Immunol. 2020, 11, 1337. [Google Scholar] [CrossRef]
- Pehote, G.; Vij, N. Autophagy Augmentation to Alleviate Immune Response Dysfunction, and Resolve Respiratory and COVID-19 Exacerbations. Cells 2020, 9, 1952. [Google Scholar] [CrossRef]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef]
- Brest, P.; Benzaquen, J.; Klionsky, D.J.; Hofman, P.; Mograbi, B. Open questions for harnessing autophagy-modulating drugs in the SARS-CoV-2 war: Hope or hype? Autophagy 2020, 1–4. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: A possible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Morris, G.; Athan, E.; Walder, K.; Bortolasci, C.C.; O’Neil, A.; Marx, W.; Berk, M.; Carvalho, A.F.; Maes, M.; Puri, B.K. Can endolysosomal deacidification and inhibition of autophagy prevent severe COVID-19? Life Sci. 2020, 262, 118541. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. The mechanistic target of rapamycin (mTOR): Novel Considerations as an Antiviral Treatment and Possibilities for COVID-19. Curr. Neurovasc. Res. 2020, 17. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 2020, 585, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, Y.; Fan, X.; Wang, X.; Han, Q.; Liu, Z. Advances in the use of chloroquine and hydroxychloroquine for the treatment of COVID-19. Postgrad. Med. 2020, 132, 604–613. [Google Scholar] [CrossRef]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Disbrow, G.L. Letter Revoking EUA for Chloroquine Phosphate and Hydroxychloroquine Sulfate. Available online: https://www.fda.gov/media/138945/download (accessed on 14 June 2020).
- World Health Organization. Solidarity Clinical Trial for COVID-19 Treatments. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments (accessed on 14 June 2020).
- Recovery Collaborative Group; Horby, P.; Mafham, M.; Linsell, L.; Bell, J.L.; Staplin, N.; Emberson, J.R.; Wiselka, M.; Ustianowski, A.; Elmahi, E.; et al. Effect of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 21, 2030–2070. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Halfon, P.; Bestion, E.; Zandi, K.; Andreani, J.; Baudoin, J.-P.; La Scola, B.; Mege, J.-L.; Mezouar, S.; Schinazi, R.F. GNS561 exhibits potent in vitro antiviral activity against SARS-CoV-2 through autophagy inhibition. bioRxiv 2020. [Google Scholar] [CrossRef]
- U.S. Department of Health & Human Services. Available online: https://www.clinicaltrials.gov/ (accessed on 2 January 2021).
- Gorshkov, K.; Chen, C.; Bostwick, R.; Rasmussen, L.; Xu, M.; Pradhan, M.; Tran, B.N.; Zhu, W.; Shamim, K.; Huang, W.; et al. The SARS-CoV-2 cytopathic effect is blocked with autophagy modulators. bioRxiv 2020. [Google Scholar] [CrossRef]
- Parnham, M.J.; Haber, V.E.; Giamarellos-Bourboulis, E.J.; Perletti, G.; Verleden, G.M.; Vos, R. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol. Ther. 2014, 143, 225–245. [Google Scholar] [CrossRef]
- Gielen, V.; Johnston, S.L.; Edwards, M.R. Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur. Respir. J. 2010, 36, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Madrid, P.B.; Panchal, R.G.; Warren, T.K.; Shurtleff, A.C.; Endsley, A.N.; Green, C.E.; Kolokoltsov, A.; Davey, R.; Manger, I.D.; Gilfillan, L.; et al. Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis. 2016, 1, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.H.; Sugamata, R.; Hirose, T.; Suzuki, S.; Noguchi, Y.; Sugawara, A.; Ito, F.; Yamamoto, T.; Kawachi, S.; Akagawa, K.S.; et al. Azithromycin, a 15-membered macrolide antibiotic, inhibits influenza A(H1N1)pdm09 virus infection by interfering with virus internalization process. J. Antibiot. 2019, 72, 759–768. [Google Scholar] [CrossRef]
- Poschet, J.F.; Perkett, E.A.; Timmins, G.S.; Deretic, V. Azithromycin and ciprofloxacin have a chloroquine-like effect on respiratory epithelial cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Andreani, J.; Le, M.; Du, I.; Jardot, P.; Rolland, C. In vitro testing of combined hydroxychloroquine and azithromycin on SARS- CoV-2 shows synergistic effect. Microb. Pathog. J. 2020, 145, 104228. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Chahinian, H.; Yahi, N. Synergistic antiviral effect of hydroxychloroquine and azithromycin in combination against SARS-CoV-2: What molecular dynamics studies of virus-host interactions reveal. Int. J. Antimicrob. Agents 2020, 56, 106020. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.C.; Weaver, J.; Kostka, K.; Duarte-Salles, T.; Abrahao, M.T.F.; Alghoul, H.; Alser, O.; Alshammari, T.M.; Biedermann, P.; Burn, E.; et al. Safety of hydroxychloroquine, alone and in combination with azithromycin, in light of rapid wide-spread use for COVID-19: A multinational, network cohort and self-controlled case series study. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Million, M.; Lagier, J.; Gautret, P.; Colson, P.; Fournier, P.; Amrane, S.; Hocquart, M.; Mailhe, M. Early treatment of COVID-19 patients with hydroxychloroquine and azithromycin: A retrospective analysis of 1061 cases in Marseille, France. Travel Med. Infect. Dis. J. 2020, 35, 101738. [Google Scholar] [CrossRef]
- Molinaa, J.M.; Delaugerreb, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirte, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical ben- efit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Méd. Mal. Infect. 2020, 382–387. [Google Scholar]
- Gbinigie, K.; Frie, K. Should azithromycin be used to treat COVID-19? A rapid review. BJGP Open 2020. [Google Scholar] [CrossRef]
- Wu, R.; Wang, L.; Kuo, H.C.D.; Shannar, A.; Peter, R.; Chou, P.J.; Li, S.; Hudlikar, R.; Liu, X.; Liu, Z.; et al. An Update on Current Therapeutic Drugs Treating COVID-19. Curr. Pharmacol. Rep. 2020, 6, 56–70. [Google Scholar] [CrossRef]
- Medhi, B.; Patyar, S.; Rao, R.S.; Byrav DS, P.; Prakash, A. Pharmacokinetic and Toxicological Profile of Artemisinin Compounds: An Update. Pharmacology 2009, 84, 323–332. [Google Scholar] [CrossRef]
- Sun, X.; Yan, P.; Zou, C.; Wong, Y.; Shu, Y.; Lee, Y.M.; Zhang, C.; Yang, N.; Wang, J.; Zhang, J. Targeting autophagy enhances the anticancer effect of artemisinin and its derivatives. Med. Res. Rev. 2019, 39, 2172–2193. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Efferth, T.; Eils, R.; Brady, N.R. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Guan, Y. Artemisinin induces selective and potent anticancer effects in drug-resistant breast cancer cells by inducing cellular apoptosis and autophagy and G2/M cell cycle arrest. JBUON 2020, 25, 1330–1336. [Google Scholar]
- Meshnick, S.R. Artemisinin: Mechanisms of action, resistance and toxicity. In Proceedings of the International Journal for Parasitology. Int. J. Parasitol. 2002, 32, 1655–1660. [Google Scholar] [CrossRef]
- Efferth, T. Beyond malaria: The inhibition of viruses by artemisinin-type compounds. Biotechnol. Adv. 2018, 36, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Li, H.; Yang, Y.; Hou, L. Anti-Inflammatory and Immunoregulatory Functions of Artemisinin and Its Derivatives. Mediat. Inflamm. 2015, 2015, 435713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.S.; Li, J.; Wang, Z.; Mi, C.; Ma, J.; Piao, L.X.; Xu, G.H.; Li, X.; Jin, X. Artemisinin inhibits inflammatory response via regulating NF-κB and MAPK signaling pathways. Immunopharmacol. Immunotoxicol. 2017, 39, 28–36. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-B-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Hu, H.; Li, Y.; Wang, X.; Xu, M.; Liu, J.; Zhang, H.; Yan, Y.; Zhao, L.; Li, W.; et al. Anti-SARS-CoV-2 Potential of Artemisinins In Vitro. ACS Infect. Dis. 2020, 6, 2524–2531. [Google Scholar] [CrossRef]
- Lee, H.C.; Aarhus, R. Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J. Cell Sci. 2000, 113, 4413–4420. [Google Scholar]
- Patel, S.; Churchill, G.C.; Galione, A. Coordination of Ca2+ signalling by NAADP. Trends Biochem. Sci. 2001, 26, 482–489. [Google Scholar] [CrossRef]
- Galione, A. NAADP, a new intracellular messenger that mobilizes Ca2+ from acidic stores. Biochem. Soc. Trans. 2006, 34, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC Isoforms Form NAADP Receptors with Distinct Roles for Ca2+ Signaling and Endolysosomal Trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; Do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, G.J.S.; Hirata, H.; do Carmo, L.G.; Stilhano, R.S.; Ureshino, R.P.; Medaglia, N.C.; Han, S.W.; Churchill, G.; Bincoletto, C.; Patel, S.; et al. NAADP-sensitive two-pore channels are present and functional in gastric smooth muscle cells. Cell Calcium 2014, 56, 51–58. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Rah, S.; Lee, Y.; Kim, U. NAADP-mediated Ca2+ signaling promotes autophagy and protects against LPS-induced liver injury. FASEB J. 2017, 31, 3126–3137. [Google Scholar] [CrossRef] [Green Version]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. MTORC1 controls lysosomal Ca2+ release through the two-pore channel TPC2. Sci. Signal. 2018, 11, 5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus Glycoprotein Directs Fusion through NPC1 + Endolysosomes. J. Virol. 2016, 90, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1151–1161. [Google Scholar] [CrossRef]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Sisk, J.M.; Frieman, M.B.; Machamer, C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018, 99, 619–630. [Google Scholar] [CrossRef]
- García, M.; Cooper, A.; Shi, W.; Bornmann, W.; Carrion, R.; Kalman, D.; Nabel, G.J. Productive replication of ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci. Transl. Med. 2012, 4, 123ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, C.M.; Sisk, J.M.; Mingo, R.M.; Nelson, E.A.; White, J.M.; Frieman, M.B. Abelson Kinase Inhibitors Are Potent Inhibitors of Severe Acute Respiratory Syndrome Coronavirus and Middle East Respiratory Syndrome Coronavirus Fusion. J. Virol. 2016, 90, 8924–8933. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Habtemariam, S.; Clementi, E.; Berindan-Neagoe, I.; Cismaru, C.A.; Rasekhian, M.; Banach, M.; Izadi, M.; Bagheri, M.; Bagheri, M.S.; et al. Lessons learned from SARS-CoV and MERS-CoV: FDA-approved Abelson tyrosine-protein kinase 2 inhibitors may help us combat SARS-CoV-2. Arch. Med. Sci. 2020, 16, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ortega, A.; Bernal-Bello, D.; Llarena-Barroso, C.; Frutos-Pérez, B.; Duarte-Millán, M.Á.; de Viedma-García, V.G.; Farfán-Sedano, A.I.; Canalejo-Castrillero, E.; Ruiz-Giardin, J.M.; Ruiz-Ruiz, J.; et al. Imatinib for COVID-19: A case report. Clin. Immunol. 2020, 218, 108518. [Google Scholar] [CrossRef] [PubMed]
- Blaess, M.; Kaiser, L.; Sauer, M.; Csuk, R.; Deigner, H.P. COVID-19/SARS-CoV-2 infection: Lysosomes and lysosomotropism implicate new treatment strategies and personal risks. Int. J. Mol. Sci. 2020, 21, 4953. [Google Scholar] [CrossRef] [PubMed]
- Encinar, J.S.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2′-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Tuck, A.; Norgren, K.; Munic Kos, V. Existing highly accumulating lysosomotropic drugs with potential for repurposing to target COVID-19. Biomed. Pharmacother. 2020, 130, 110582. [Google Scholar] [CrossRef]
- Sauvat, A.; Ciccosanti, F.; Colavita, F.; Di Rienzo, M.; Castilletti, C.; Capobianchi, M.R.; Kepp, O.; Zitvogel, L.; Fimia, G.M.; Piacentini, M.; et al. On-target versus off-target effects of drugs inhibiting the replication of SARS-CoV-2. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, L.; Kantarjian, H. Targeted therapy: Generic imatinib-impact on frontline and salvage therapy for CML. Nat. Rev. Clin. Oncol. 2016, 13, 270–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joensuu, H.; Dematteo, R.P. The management of gastrointestinal stromal tumors: A model for targeted and multidisciplinary therapy of malignancy. Annu. Rev. Med. 2012, 63, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef] [PubMed]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Barlow, A.; Landolf, K.M.; Barlow, B.; Yeung, S.Y.A.; Heavner, J.J.; Claassen, C.W.; Heavner, M.S. Review of Emerging Pharmacotherapy for the Treatment of Coronavirus Disease 2019. Pharmacotherapy 2020, 40, 416–437. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.W.; Yao, Y.; Yeung, M.L.; Deng, W.; Bao, L.; Jia, L.; Li, F.; Xiao, C.; Gao, H.; Yu, P.; et al. Treatment with lopinavir/ritonavir or interferon-β1b improves outcome of MERSCoV infection in a nonhuman primate model of common marmoset. J. Infect. Dis. 2015, 212, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Boriskin, Y.S.; Leneva, I.A.; Pécheur, E.-I.; Polyak, S.J. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef]
- Boriskin, Y.S.; Pécheur, E.I.; Polyak, S.J. Arbidol: A broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virol. J. 2006, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallam, H.J.; Hallam, S.; Rodriguez, S.E.; Barrett, A.D.T.; Beasley, D.W.C.; Chua, A.; Ksiazek, T.G.; Milligan, G.N.; Sathiyamoorthy, V.; Reece, L.M. Baseline mapping of Lassa fever virology, epidemiology and vaccine research and development review-article. NPJ Vaccines 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Hulseberg, C.E.; Fénéant, L.; Szyman’ska, K.M.; Szyman’ska-De Wijs, S.; Kessler, N.P.; Nelson, E.A.; Shoemaker, C.J.; Schmaljohn, C.S.; Polyak, S.J.; White, J.M.; et al. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses Downloaded from. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaising, J.; Polyak, S.J.; Pécheur, E.I. Arbidol as a broad-spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide antibiotics potently inhibit cathepsin l in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ma, X.; Yu, F.; Liu, J.; Zou, F.; Pan, T.; Zhang, H. Teicoplanin potently blocks the cell entry of 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Pandrea, I.; Landay, A.L. Implications for Therapy. In Models of Protection Against HIV/SIV; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 81–132. ISBN 9780123877154. [Google Scholar]
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.C.; Chu, H.; Han, S.H.; Yun, C.H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95. [Google Scholar] [CrossRef]
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L.; et al. Autophagy Negatively Regulates Transmissible Gastroenteritis Virus Replication. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, C.; Wong, W.; Mak, L.; Wang, X.; Chu, H.; Yuen, K.; Kok, K. Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J. Med. Virol. 2020, jmv.26583. [Google Scholar] [CrossRef] [PubMed]
- Appelberg, S.; Gupta, S.; Akusjärvi, S.S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Basu, A.; Kanda, T.; Beyene, A.; Saito, K.; Meyer, K.; Ray, R. Sulfated Homologues of Heparin Inhibit Hepatitis C Virus Entry into Mammalian Cells. J. Virol. 2007, 81, 3933–3941. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, S.; Cooper, L.; Rubio, A.; Pagani, I.; Rosaria, M.; Ippolito, G.; Pelletier, J.; Yates, E.A.; Vicenzi, E. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells Silvia. Antivir. Res. 2020, 140, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Vicenzi, E.; Canducci, F.; Pinna, D.; Mancini, N.; Carletti, S.; Lazzarin, A.; Bordignon, C.; Poli, G.; Clementi, M. Coronaviridae and SARS-associated Coronavirus Strain HSR1. Emerg. Infect. Dis. 2004, 10, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Guimond, S.E.; Miller, G.; Meneghetti, M.C.Z.; Nader, H.B.; Li, Y.; et al. Heparin inhibits cellular invasion by SARS-CoV-2: Structural dependence of the interaction of the surface protein (spike) S1 receptor binding domain with heparin. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.C.; Heimann, M.A.; Burleson, S.L.; Anzalone, B.C.; Swanson, J.F.; Wallace, D.W.; Greene, C.J. COVID-19: A review of therapeutics under investigation. J. Am. Coll. Emerg. Physicians Open 2020, 1, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Favalli, E.G.; Ingegnoli, F.; Cimaz, R.; Caporali, R.; DeLucia, O.; Cincinelli, G. COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun. Rev. J. 2020, 19, 102523. [Google Scholar] [CrossRef] [PubMed]
- Yamaya, M.; Nishimura, H.; Deng, X.; Sugawara, M.; Watanabe, O.; Nomura, K.; Shimotai, Y.; Momma, H.; Ichinose, M.; Kawase, T. Inhibitory effects of glycopyrronium, formoterol, and budesonide on coronavirus HCoV-229E replication and cytokine production by primary cultures of human nasal and tracheal epithelial cells. Respir. Investig. 2020, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Song, J.H.; Ahn, J.H.; Lee, G.S.; Ahn, H.; Yoon, S.-I.; Kang, S.G.; Kim, P.H.; Jeon, S.M.; Choi, E.J.; et al. Antiviral and anti-inflammatory activity of budesonide against human rhinovirus infection mediated via autophagy activation. Antivir. Res. 2018, 151, 87–96. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Song, J.; Lee, Y.; Ko, H. Anti-Human Rhinovirus 1B Activity of Dexamethasone via GCR-Dependent Autophagy Activation Jae-Sug. Osong Public Health Res. Perspect. 2012, 9, 334. [Google Scholar] [CrossRef]
- He, Q.; Song, X.; Huang, Y.; Huang, W.; Ye, B.; Luo, H.; Luo, H.; Wu, L.; Wang, Z.; Chen, W.X.; et al. Dexamethasone stimulates hepatitis B virus (HBV) replication through autophagy. Med. Sci. Monit. 2018, 24, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, H.; Yang, F.; Wang, Q.; Feng, Y.; Zhang, C. Glucocorticoid-activated IRE1α/XBP-1s signaling: An autophagy-associated protective pathway against endotheliocyte damage. Am. J. Physiol. Cell Physiol. 2018, 315, C300–C309. [Google Scholar] [CrossRef]
- Ahn, S.; Park, J.; An, I.; Jung, S.J.; Hwang, J. Transient Receptor Potential cation channel V1 (TRPV1) is degraded by starvation- and glucocorticoid-mediated autophagy. Mol. Cells 2014, 37, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncoso, R.; Paredes, F.; Parra, V.; Gatica, D.; Vásquez-Trincado, C.; Quiroga, C.; Bravo-Sagua, R.; López-Crisosto, C.; Rodriguez, A.E.; Oyarzuń, A.P.; et al. Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle 2014, 13, 2281–2295. [Google Scholar] [CrossRef] [Green Version]
- Kar, R.; Riquelme, M.A.; Hua, R.; Jiang, J.X. Glucocorticoid-Induced Autophagy Protects Osteocytes Against Oxidative Stress Through Activation of MAPK/ERK Signaling. JBMR Plus 2019, 3, e10077. [Google Scholar] [CrossRef]
- Juszczak, G.R.; Stankiewicz, A.M. Glucocorticoids, genes and brain function. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 136–168. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019, 5, 8857–8865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, M.C.; Sajuthi, S.; Deford, P.; Christenson, S.; Rios, C.L.; Montgomery, M.T.; Woodruff, P.G.; Mauger, D.T.; Erzurum, S.C.; Johansson, M.W.; et al. COVID-19-related genes in sputum cells in asthma: Relationship to demographic features and corticosteroids. Am. J. Respir. Crit. Care Med. 2020, 202, 83–90. [Google Scholar] [CrossRef]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA 2020, 323, 2427. [Google Scholar] [CrossRef]
- Cattrini, C.; Bersanelli, M.; Latocca, M.M.; Conte, B.; Vallome, G.; Boccardo, F. Sex hormones and hormone therapy during covid-19 pandemic: Implications for patients with cancer. Cancers 2020, 12, 2325. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Pfeffer, M.A. Plasma angiotensin-converting enzyme 2: Novel biomarker in heart failure with implications for COVID-19. Eur. Heart J. 2020, 41, 1818–1820. [Google Scholar] [CrossRef] [PubMed]
- Sama, I.E.; Ravera, A.; Santema, B.T.; Van Goor, H.; Ter Maaten, J.M.; Cleland, J.G.F.; Rienstra, M.; Friedrich, A.W.; Samani, N.J.; Ng, L.L.; et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 inmen and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur. Heart J. 2020, 41, 1810–1817. [Google Scholar] [CrossRef]
- Piccinni, M.P.; Giudizi, M.G.; Biagiotti, R.; Beloni, L.; Giannarini, L.; Sampognaro, S.; Parronchi, P.; Manetti, R.; Annunziato, F.; Livi, C. Progesterone favors the development of human T helper cells producing Th2-type cytokines and promotes both IL-4 production and membrane CD30 expression in established Th1 cell clones. J. Immunol. 1995, 155, 128–133. [Google Scholar] [PubMed]
- Stilhano, R.S.; Costa, A.J.; Nishino, M.S.; Shams, S.; Bartolomeo, C.S.; Breithaupt-Faloppa, A.C.; Silva, E.A.; Ramirez, A.L.; Prado, C.M.; Ureshino, R.P. SARS-CoV-2 and the possible connection to ERs, ACE2, and RAGE: Focus on susceptibility factors. FASEB J. 2020, 34, 14103–14119. [Google Scholar] [CrossRef]
- Stelzig, K.E.; Canepa-Escaro, F.; Schiliro, M.; Berdnikovs, S.; Prakash, Y.S.; Chiarella, S.E. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Mariana, R.; Lemes, R.; Jardim, A.; Cynthia, C.; Bartolomeo, S.; Bassani, T.B.; Nishino, M.S.; Jose, G.; Pereira, S.; Soubhi, S.; et al. 17β-estradiol reduces SARS-CoV-2 infection in vitro. Physiol. Rep. 2021, 9, e14707. [Google Scholar] [CrossRef]
- Pozzilli, P.; Lenzi, A. Testosterone, a key hormone in the context of COVID-19 pandemic. Metabolism 2020, 108, 154252. [Google Scholar] [CrossRef]
- Salonia, A.; Corona, G.; Giwercman, A.; Maggi, M.; Minhas, S.; Nappi, R.E.; Sofikitis, N.; Vignozzi, L. SARS-CoV-2, testosterone and frailty in males (PROTEGGIMI): A multidimensional research project. Andrology 2020, 9, 19–22. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and androgen-regulated expression of the membrane- bound serine protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [Green Version]
- Stopsack, K.H.; Mucci, L.A.; Antonarakis, E.S.; Nelson, P.S.; Kantoff, P.W. TMPRSS2 and COVID-19: Serendipity or opportunity for intervention? Cancer Discov. 2020, 10, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Phil, D.; Sta-Plin, N.; Brightling, C.; Med, F.; et al. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Guo, J.; Zou, Z.; Liu, J.; Cao, B.; Zhang, S.; Li, H.; Wang, W.; Sheng, M.; Liu, S.; et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat. Commun. 2014, 5, 3595. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, F.; Xu, J.; Yang, P.; Qin, Y.; Cao, M.; Wang, Z.; Li, X.; Zhang, S.; Ye, L.; et al. Anti-hypertensive Angiotensin II receptor blockers associated to mitigation of disease severity in elderly COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 1–5. [Google Scholar] [CrossRef]
- Porrello, E.R.; D’Amore, A.; Curl, C.L.; Allen, A.M.; Harrap, S.B.; Thomas, W.G.; Delbridge, L.M.D. Angiotensin II Type 2 Receptor Antagonizes Angiotensin II Type 1 Receptor–Mediated Cardiomyocyte Autophagy. Hypertension 2009, 53, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrello, E.R.; Delbridge, L.M.D. Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy 2009, 5, 1215–1216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, J.; Yan, Y.; Ruan, Z.; Su, Y.; Wang, J.; Huang, H.; Zhang, Y.; Wang, W.; Gao, J.; et al. Angiotensin-converting enzyme 2 regulates autophagy in acute lung injury through AMPK/mTOR signaling. Arch. Biochem. Biophys. 2019, 672, 108061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality among Patients with Hypertension Hospitalized with COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Harvey, D.J.; Samara, E.; Mechoulam, R. Comparative metabolism of cannabidiol in dog, rat and man. Pharmacol. Biochem. Behav. 1991, 40, 523–532. [Google Scholar] [CrossRef]
- Malfait, A.M.; Gallily, R.; Sumariwalla, P.F.; Malik, A.S.; Andreakos, E.; Mechoulam, R.; Feldmann, M. The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 9561–9566. [Google Scholar] [CrossRef] [Green Version]
- Capasso, R.; Borrelli, F.; Aviello, G.; Romano, B.; Scalisi, C.; Capasso, F.; Izzo, A.A. Cannabidiol, extracted from Cannabis sativa, selectively inhibits inflammatory hypermotility in mice. Br. J. Pharmacol. 2008, 154, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; de Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Ribeiro, A.; Almeida, V.I.; Costola-De-Souza, C.; Ferraz-De-Paula, V.; Pinheiro, M.L.; Vitoretti, L.B.; Gimenes-Junior, J.A.; Akamine, A.T.; Crippa, J.A.; Tavares-De-Lima, W.; et al. Cannabidiol improves lung function and inflammation in mice submitted to LPS-induced acute lung injury. Immunopharmacol. Immunotoxicol. 2015, 37, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.; Shahinas, J. Dosage, Efficacy and Safety of Cannabidiol Administration in Adults: A Systematic Review of Human Trials. J. Clin. Med. Res. 2020, 12, 129–141. [Google Scholar] [CrossRef]
- Costiniuk, C.T.; Jenabian, M.A. Acute inflammation and pathogenesis of SARS-CoV-2 infection: Cannabidiol as a potential anti-inflammatory treatment? Cytokine Growth Factor Rev. 2020, 53, 63–65. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013. [Google Scholar] [CrossRef]
- Kozela, E.; Pietr, M.; Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z. Cannabinoids Δ9-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-κB and interferon-β/STAT proinflammatory pathways in BV-2 microglial cells. J. Biol. Chem. 2010, 285, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Vuolo, F.; Petronilho, F.; Sonai, B.; Ritter, C.; Hallak, J.E.C.; Zuardi, A.W.; Crippa, J.A.; Dal-Pizzol, F. Evaluation of Serum Cytokines Levels and the Role of Cannabidiol Treatment in Animal Model of Asthma. Mediat. Inflamm. 2015, 2015. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Byrareddy, S.N.; Mohan, M. SARS-CoV2 induced respiratory distress: Can cannabinoids be added to anti-viral therapies to reduce lung inflammation? Brain Behav. Immun. 2020, 87, 120. [Google Scholar] [CrossRef]
- Wang, B.; Kovalchuk, A.; Li, D.; Ilnytskyy, Y.; Kovalchuk, I. In Search of Preventative Strategies: Novel Anti-Inflammatory High-CBD Cannabis Sativa Extracts Modulate ACE2 Expression in COVID-19 Gateway Tissues. Preprints 2020, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Machado Bergamaschi, M.; Helena Costa Queiroz, R.; Waldo Zuardi, A.; Alexandre, S.; Crippa, J. Safety and Side Effects of Cannabidiol, a Cannabis sativa Constituent. Curr. Drug Saf. 2011, 6, 237–249. [Google Scholar] [CrossRef]
- Huang, S.; Goplen, N.P.; Zhu, B.; Cheon, I.S.; Son, Y.; Wang, Z.; Li, C.; Dai, Q.; Jiang, L.; Xiang, M.; et al. Macrophage PPAR-γ suppresses long-term lung fibrotic sequelae following acute influenza infection. PLoS ONE 2019, 14, e223430. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.; Ferraz-De-Paula, V.; Pinheiro, M.L.; Vitoretti, L.B.; Mariano-Souza, D.P.; Quinteiro-Filho, W.M.; Akamine, A.T.; Almeida, V.I.; Quevedo, J.; Dal-Pizzol, F.; et al. Cannabidiol, a non-psychotropic plant-derived cannabinoid, decreases inflammation in a murine model of acute lung injury: Role for the adenosine A 2A receptor. Eur. J. Pharmacol. 2012, 678, 78–85. [Google Scholar] [CrossRef]
- Costa, L.; Amaral, C.; Teixeira, N.; Correia-da-Silva, G.; Fonseca, B.M. Cannabinoid-induced autophagy: Protective or death role? Prostaglandins Other Lipid Mediat. 2016, 122, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Koay, L.C.; Rigby, R.J.; Wright, K.L. Cannabinoid-induced autophagy regulates suppressor of cytokine signaling-3 in intestinal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, 140–148. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Tsuchiya, Y.; Nakayama, K.; Satoh, T.; Nishida, E. Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells 2008, 13, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Hiebel, C.; Kromm, T.; Stark, M.; Behl, C. Cannabinoid receptor 1 modulates the autophagic flux independent of mTOR- and BECLIN1-complex. J. Neurochem. 2014, 131, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.L.; Jackson, W.T. That which does not degrade you makes you stronger. Autophagy 2013, 9, 806–807. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Rahman, M.A.; Rahman, M.S.; Sohag, A.A.M.; Dash, R.; Hossain, K.S.; Farjana, M.; Uddin, M.J. Intermittent fasting, a possible priming tool for host defense against SARS-CoV-2 infection: Crosstalk among calorie restriction, autophagy and immune response. Immunol. Lett. 2020, 226, 38–45. [Google Scholar] [CrossRef]
- Johnson, J.B.; Summer, W.; Cutler, R.G.; Martin, B.; Hyun, D.H.; Dixit, V.D.; Pearson, M.; Nassar, M.; Tellejohan, R.; Maudsley, S.; et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 2007, 42, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Calender, A.; Israel-Biet, D.; Valeyre, D.; Pacheco, Y. Modeling Potential Autophagy Pathways in COVID-19 and Sarcoidosis. Trends Immunol. 2020, 41, 856–859. [Google Scholar] [CrossRef]
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjós, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omarjee, L.; Janin, A.; Perrot, F.; Laviolle, B.; Meilhac, O.; Mahe, G. Targeting T-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin. Immunol. 2020, 216, 108464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Uzun, T.; Toptas, O. Artesunate: Could be an alternative drug to chloroquine in COVID-19 treatment? Chin. Med. 2020, 15, 54. [Google Scholar] [CrossRef]
- Khan, N.; Halcrow, P.W.; Lakpa, K.L.; Afghah, Z.; Miller, N.M.; Dowdy, S.F.; Geiger, J.D.; Chen, X. Two-pore channels regulate Tat endolysosome escape and Tat-mediated HIV-1 LTR transactivation. FASEB J. 2020, 34, 4147–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzorno, A.; Padey, B.; Dubois, J.; Julien, T.; Traversier, A.; Dulière, V.; Brun, P.; Lina, B.; Rosa-Calatrava, M.; Terrier, O. In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antivir. Res. 2020, 181, 104878. [Google Scholar] [CrossRef]
- Kang, C.K.; Seong, M.W.; Choi, S.J.; Kim, T.S.; Choe, P.G.; Song, S.H.; Kim, N.J.; Park, W.B.; Oh, M.D. In vitro activity of lopinavir/ritonavir and hydroxychloroquine against severe acute respiratory syndrome coronavirus 2 at concentrations achievable by usual doses. Korean J. Intern. Med. 2020, 35, 782–787. [Google Scholar] [CrossRef]
- Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.H.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef]
- Chu, C.M.; Cheng, V.C.C.; Hung, I.F.N.; Wong, M.M.L.; Chan, K.H.; Chan, K.S.; Kao, R.Y.T.; Poon, L.L.M.; Wong, C.L.P.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, A.H.; Jochmans, D.; Posthuma, C.C.; Zevenhoven-Dobbe, J.C.; Van Nieuwkoop, S.; Bestebroer, T.M.; Van Den Hoogen, B.G.; Neyts, J.; Snijder, E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014, 58, 4875–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Manuzak, J.A.; Gott, T.M.; Kirkwood, J.S.; Coronado, E.; Hensley-Mcbain, T.; Miller, C.; Cheu, R.K.; Collier, A.C.; Funderburg, N.T.; Martin, J.N.; et al. Heavy Cannabis Use Associated with Reduction in Activated and Inflammatory Immune Cell Frequencies in Antiretroviral Therapy-Treated Human Immunodeficiency Virus-Infected Individuals. Clin. Infect. Dis. 2018, 66, 1872–1882. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, M.D.; Crawford, R.B.; Henriquez, J.E.; Aldhamen, Y.; Gulick, P.; Amalfitano, A.; Kaminski, N.E. HIV-infected cannabis users have lower circulating CD16+ monocytes and IP-10 levels compared to non-using HIV patients. AIDS 2017, 32, 419. [Google Scholar] [CrossRef]
- National Library of Medicine National Center for Biotechnology Information. PubChem Database. Explore Chemistry. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 2 January 2021).



{kind=link}
{kind=link}
{kind=link}
| Acronym | Protein | Function | Ref. |
|---|---|---|---|
| 1. Transcriptional factors | |||
| TFEB | Transcription factor EB | A master gene regulator of lysosomal biogenesis and autophagy | [54,55] |
| 2. Initiation of autophagy | |||
| mTORC1 | Mammalian target of rapamycin complex 1 | Nutrient sensor and controller of protein synthesis and autophagy | [34] |
| 3. Upstream regulators of mTORC1 | |||
| AKT | Serine-threonine kinase | Cell growth, proliferation, differentiation and survival signalling | [35,36] |
| AMPK | Adenosine monophosphate-activated protein kinase | Energy homeostasis signalling | [37] |
| BCL-2 | B-cell lymphoma 2 | Regulation of cell death | [40,41] |
| ERK/MAPK | Extracellular signal-regulated kinase/mitogen-activated protein kinase | Regulation of cell proliferation | [61] |
| PI3K | Phosphoinositide 3-kinase | Cell growth, proliferation, differentiation and survival signalling | [35,36] |
| 4. Nucleation and phagophore formation | |||
| Ambra1 | Activating molecule in Beclin-1-regulated autophagy | Positive regulator of Beclin-1-mediated autophagy | [42] |
| BECN1 | Beclin-1 | Regulator of autophagic programmed cell death | [40,41] |
| ULK1 | Unc-51 like autophagy activating kinase | Autophagy initiator | [38,39] |
| 5. Autophagosome formation and elongation | |||
| Atg | Autophagy-related protein | Factors required for the formation of autophagosomal membranes | [34] |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3A | Autophagosomal marker that mediates the physical interactions between microtubules and components of the cytoskeleton | [43] |
| p62/SQSTM1 | Ubiquitin-binding protein p62/Sequestosome-1 | An autophagosome cargo protein that targets and labels other proteins for selective autophagy | [56,70] |
| Vps34 | Vacuolar protein sorting 34 | A class III phosphoinositide 3-kinase that acts on vesicle trafficking | [43] |
| WIPI2 | WD repeat domain phosphoinositide-interacting protein proteins | Regulates the assembly of multiprotein complexes | [43] |
| 6. Autophagosome-lysosome fusion | |||
| SNAP29 | Synaptosome-associated protein 29 | Mediates autophagosome-lysosome fusion | [46] |
| SNARE | N-ethylmaleimide-sensitive factor attachment protein receptor complexes | Vesicle fusion mediator | [46,70] |
| Stx17 | Syntaxin 17 | A SNARE like protein that mediates autophagosome-lysosome fusion | [46,70] |
| VAMP8 | Vesicle-associated membrane protein 8 | A SNARE like protein that mediates autophagosome-lysosome fusion | [46] |
| Drug | Mechanisms | Activity (In Vitro) | Cell Model | Ref. | Current Clinical Trials Number/Phase |
|---|---|---|---|---|---|
| 1. Lysosomotropic agents | |||||
| Chloroquine/ hydroxychloroquine | -Prevents endolysosomal acidification; -Blockade of cathepsin activity; -Intracellular retention of ACE2. | SARS-CoV-2 | Vero E6 | [268] | NCT04341727/Phase 3 NCT04328272/Phase 3 |
| SARS-CoV-2 | Vero E6 | [110] | |||
| SARS-CoV | HEK293E; Vero E6 | [117] | |||
| SARS-CoV | Vero E6 | [106] | |||
| Azithromycin | -Acidotropic lipophilic weak base with similar effects to CQ in vitro; -Possible synergy with HCQ for competitive inhibition of SARS-CoV-2 attachment to the host-cell membrane; -Blockade of viral internalization in the early phase of viral infections; -Increases the production of interferon-stimulated genes in rhinoviral infections. | SARS-CoV-2 | Vero E6/ In silico | [127,128] |
NCT04321278/Phase 3 NCT04381962/Phase 3 |
| SARS-CoV-2 (presumed) | IB3-1 | [126] | |||
| H1N1 | A549 | [125] | |||
| ZIKV | Vero, U87 | [124] | |||
| EBOV | Vero E6 | [123] | |||
| HRV | HBECs | [122] | |||
| Artemisinin and its derivative compounds | -Inhibition of NF-κB; -Chloroquine-like endocytosis inhibition mechanism. | SARS-CoV-2 | Vero E6 | [144,269] | NCT04387240/Phase 2 NCT04382040/Phase 2 |
| Tetrandrine and ned-19 | -Pharmacological inhibition of TPCs; -Inhibition of viral translocation and motility in the endosomal system. | EBOV | HeLa | [158] | NCT04308317/Phase 4 |
| MERS-CoV | Huh7 | [160] | |||
| HIV-1 | U87MG | [270] | |||
| Imatinib | -Inhibitor of ABL-2; -Inhibition of the virion fusion at the endosomal membrane. | SARS-CoV, IBV | Vero E6 | [162] | NCT04422678/Phase 3 NCT04346147/Phase 4 |
| SARS-CoV, MERS-CoV | Vero E6, MRC5, Calu-3, Huh7, BSC1 | [164] | |||
| 2. Protease inhibitors/Antiviral agents | |||||
| Camostat mesylate | -Prevents the viral entrance on host cell; -Inhibition of TMPRSS2, a serine protease that cleaves the spike S protein after viral bound to ACE2 receptor. | SARS-CoV-2 | Calu-3 and Vero | [101] | NCT04338906/Phase 4 NCT04353284/Phase 2 |
| SARS-CoV | Caco2 | [175] | |||
| Lopinavir/ ritonavir | -Protease inhibitor that prevent viral replication and spread; -Inhibition of protease type 3C; Ritonavir inhibits CYP450. | SARS-CoV-2 | Vero E6 | [271] | NCT04307693/Phase 2 NCT04372628/Phase 2 |
| SARS-CoV-2 | Vero E6 | [272] | |||
| SARS-CoV-2 | Vero E6 | [273] | |||
| SARS-CoV | FRhK-4 | [274] | |||
| MERS-CoV | Vero, Huh7 | [275] | |||
| Umifenovir | -Prevents the viral invasion of host cell binding to membrane lipids; -Binds to membrane proteins like clathrin, preventing viral endocytosis through clathrin receptors. | SARS-CoV-2 | Vero E6 | [271] | NCT04476719/Phase 4 NCT04260594/Phase 4 |
| LASV, EBOV | HEK293/17 and BSC-1 | [182] | |||
| Teicoplanin and others | -Reduces viral invasion by inhibition of cathepsin L activity. | EBOV MERS-CoV SARS-CoV | HEK293, A549 and HeLa | [184] | IRCT20161204031229N3/Phase 3 |
| SARS-CoV-2 | HEK293 and Huh7 | [185] | |||
| 3. PI3K/AKT/mTOR modulators | |||||
| Rapamycin | -Inhibition of mTOR pathway; | PEDV | IPEC-J2 | [187] | NCT04482712/ Phase 1 and 2 |
| TGEV | ST, PK15 | [188] | |||
| MERS-CoV | Huh7 | [61] | |||
| Wortmannin | -Phosphatidylinositol 3-kinase (PI3K) pathway inhibition; | TGEV | ST, PK15 | [188] | N/A |
| MERS-CoV | Huh7 | [61] | |||
| Heparin | -Inhibition of viral binding with glycosaminoglycans present on the cell surface; | SARS-CoV-2 | Vero E6 | [197] | NCT04530578/Phase 4 |
| SARS-CoV | Vero E6 | [194] | |||
| SARS-CoV | HEK293E/ACE2-Myc, Vero E6, Caco-2 | [196] | |||
| HCV | IHH | [192] | |||
| Glucocorticoids | -Glucocorticoid receptor-dependent autophagy activation; -Inhibition of IL-1β, IL-6, IL-8 NF-κB, IFN-β, IFN-λ1 and IFN-γ mediated inflammation; | HCoV-229E | HNE, HTE | [205] | NCT04438980/Phase 3 |
| HRV | HeLa, Vero E6 | [206] | |||
| Losartan | -Inhibition of the AT1 receptor; | SARS-CoV | Mice (in vivo) | [276] | NCT04335123/Phase 1 |
| Cannabidiol | -Inhibition of the transmigration of blood leukocytes; -Downregulation of the vascular cell adhesion molecule-1 (VCAM-1), chemokines (CCL2 and CCL5) and the proinflammatory cytokine IL-1β expression; -Attenuation of microglial activation. | HIV | Human (in vivo) | [277,278] | NCT04467918/ Phase 2 and 3 |
| TMEV | Mice (in vivo) | [247] | |||
| Drug | Therapeutic Properties | Toxicological Properties | Compound ID (CID) |
|---|---|---|---|
| Chloroquine/hydroxycloroquine |
|
| 2719 3652 |
| Azithromicin |
|
| 447043 |
| Artemisinin |
|
| 68827 |
| Tetrandrine |
|
| 73078 |
| Imatinib |
|
| 5291 |
| Camostat mesylate |
|
| 5284360 |
| Lopinavir/ritonavir |
|
| 11979606 |
| Umifenovir |
|
| 131411 |
| Teicoplanin |
|
| 133065662 |
| Rapamycin |
|
| 5284616 |
| Heparin |
|
| 772 |
| Dexamethasone (glucocorticoid) |
|
| 5743 |
| Losartan |
|
| 3961 |
| Cannabidiol |
|
| 644019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, G.J.d.S.; Leão, A.H.F.F.; Erustes, A.G.; Morais, I.B.d.M.; Vrechi, T.A.d.M.; Zamarioli, L.d.S.; Pereira, C.A.S.; Marchioro, L.d.O.; Sperandio, L.P.; Lins, Í.V.F.; et al. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 4067. https://doi.org/10.3390/ijms22084067
Pereira GJdS, Leão AHFF, Erustes AG, Morais IBdM, Vrechi TAdM, Zamarioli LdS, Pereira CAS, Marchioro LdO, Sperandio LP, Lins ÍVF, et al. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. International Journal of Molecular Sciences. 2021; 22(8):4067. https://doi.org/10.3390/ijms22084067
Chicago/Turabian StylePereira, Gustavo José da Silva, Anderson Henrique França Figueredo Leão, Adolfo Garcia Erustes, Ingrid Beatriz de Melo Morais, Talita Aparecida de Moraes Vrechi, Lucas dos Santos Zamarioli, Cássia Arruda Souza Pereira, Laís de Oliveira Marchioro, Letícia Paulino Sperandio, Ísis Valeska Freire Lins, and et al. 2021. "Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19" International Journal of Molecular Sciences 22, no. 8: 4067. https://doi.org/10.3390/ijms22084067
APA StylePereira, G. J. d. S., Leão, A. H. F. F., Erustes, A. G., Morais, I. B. d. M., Vrechi, T. A. d. M., Zamarioli, L. d. S., Pereira, C. A. S., Marchioro, L. d. O., Sperandio, L. P., Lins, Í. V. F., Piacentini, M., Fimia, G. M., Reckziegel, P., Smaili, S. S., & Bincoletto, C. (2021). Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. International Journal of Molecular Sciences, 22(8), 4067. https://doi.org/10.3390/ijms22084067