
S100A4 Is Involved in Stimulatory Effects Elicited by the FGF2/FGFR1 Signaling Pathway in Triple-Negative Breast Cancer (TNBC) Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Gene Expression Levels of FGF2 and S100A4 Are Correlated in TNBC Patients
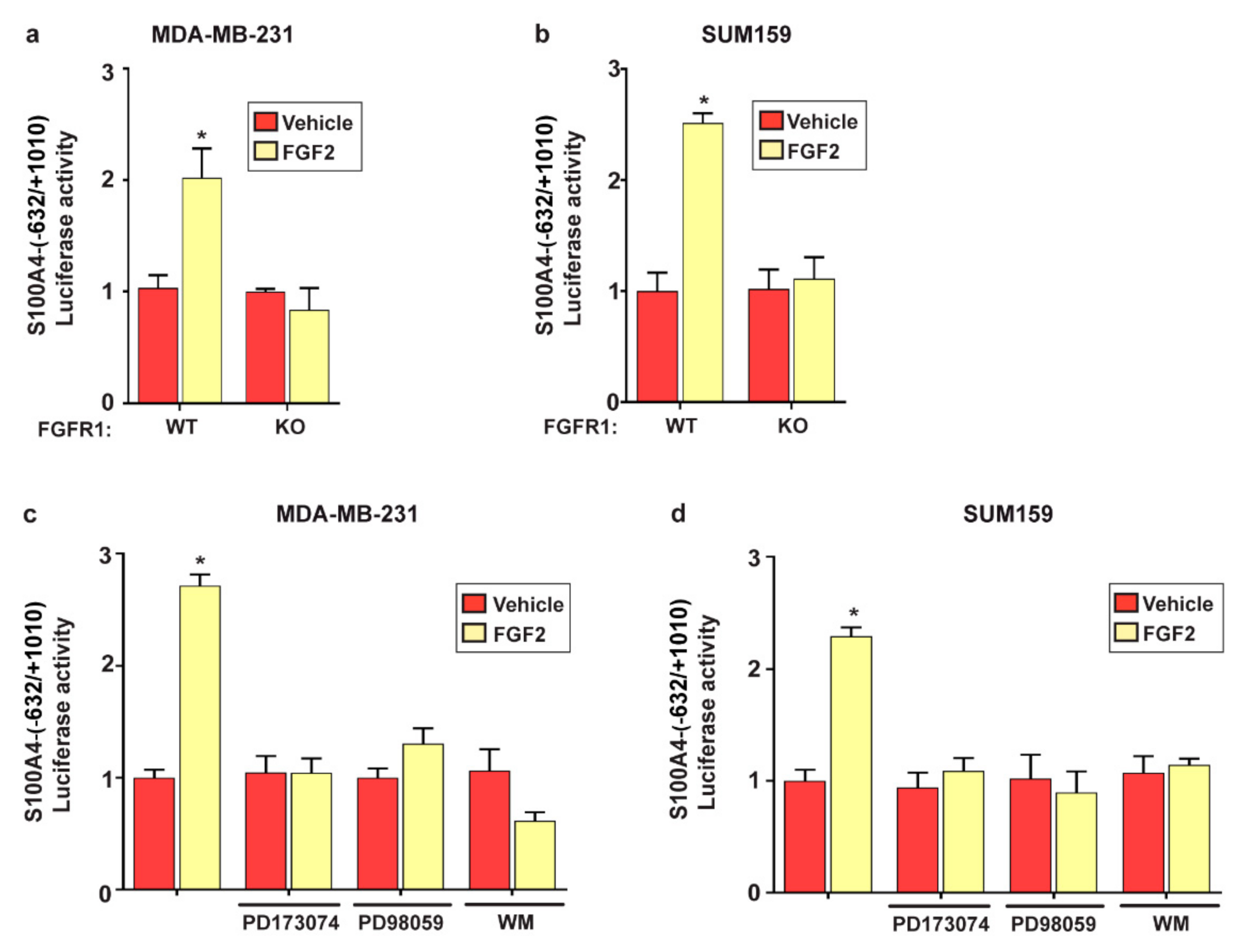
2.2. FGF2/FGFR1 Mediated Signaling Upregulates S100A4 Levels in TNBC Cells
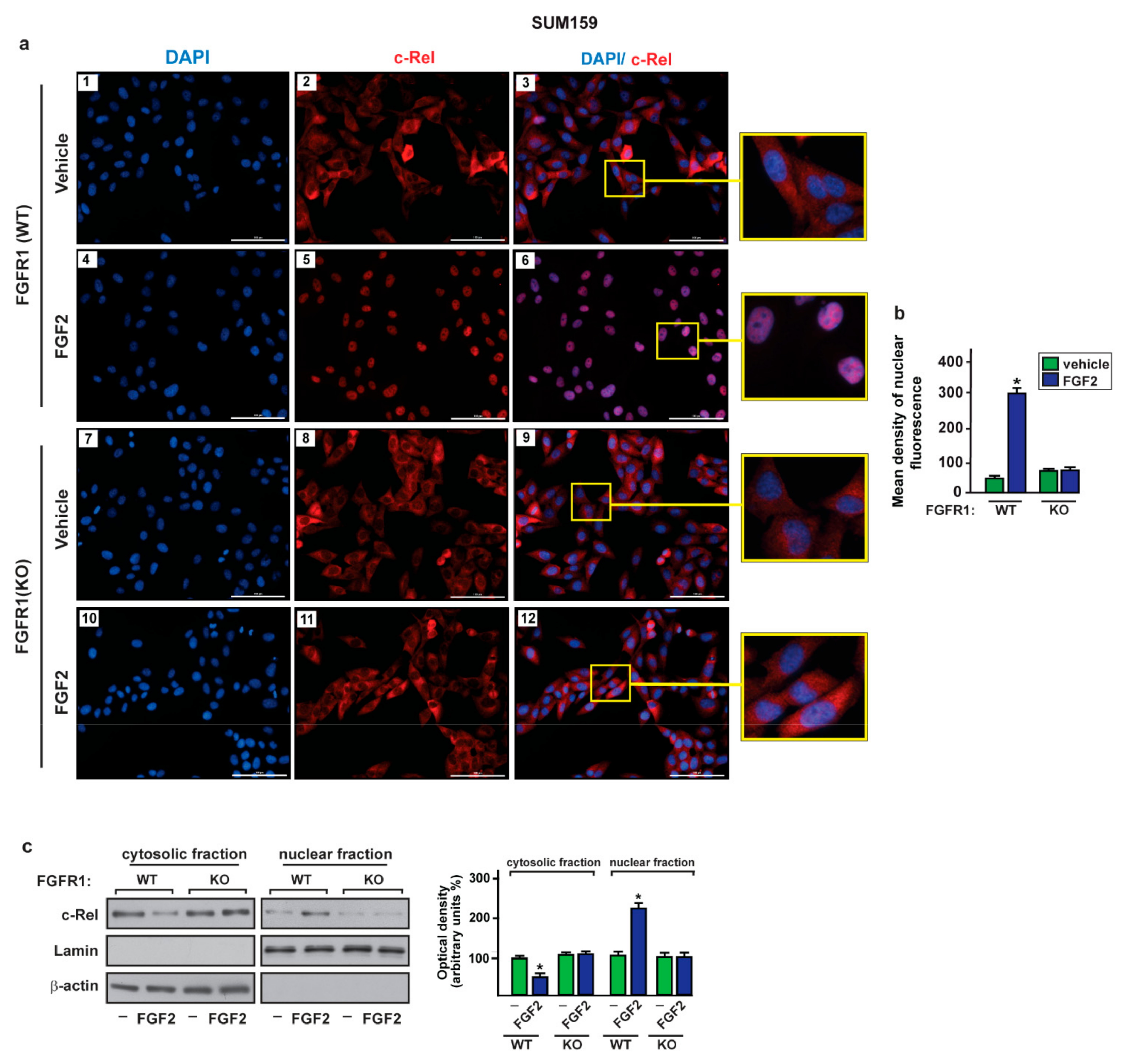
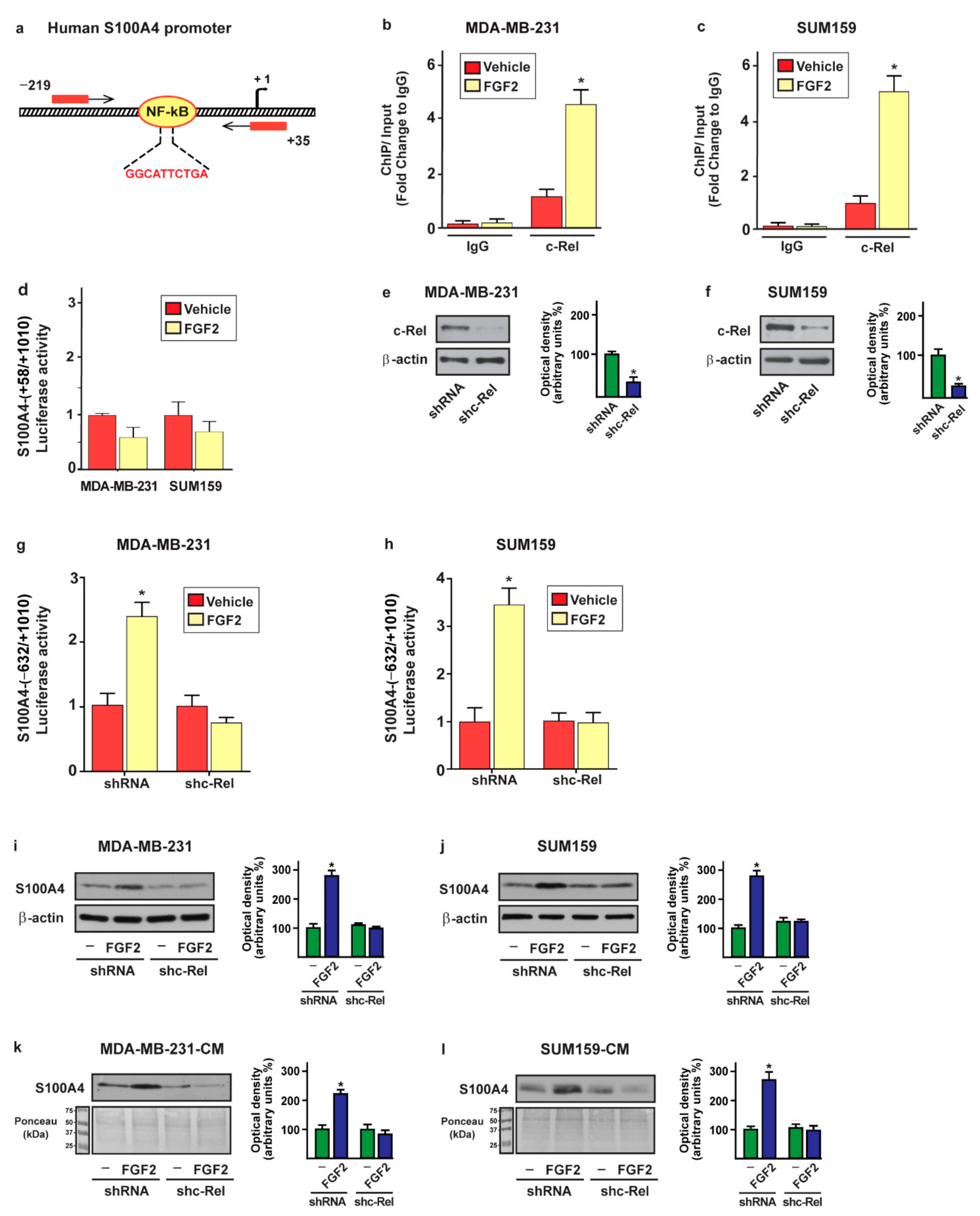
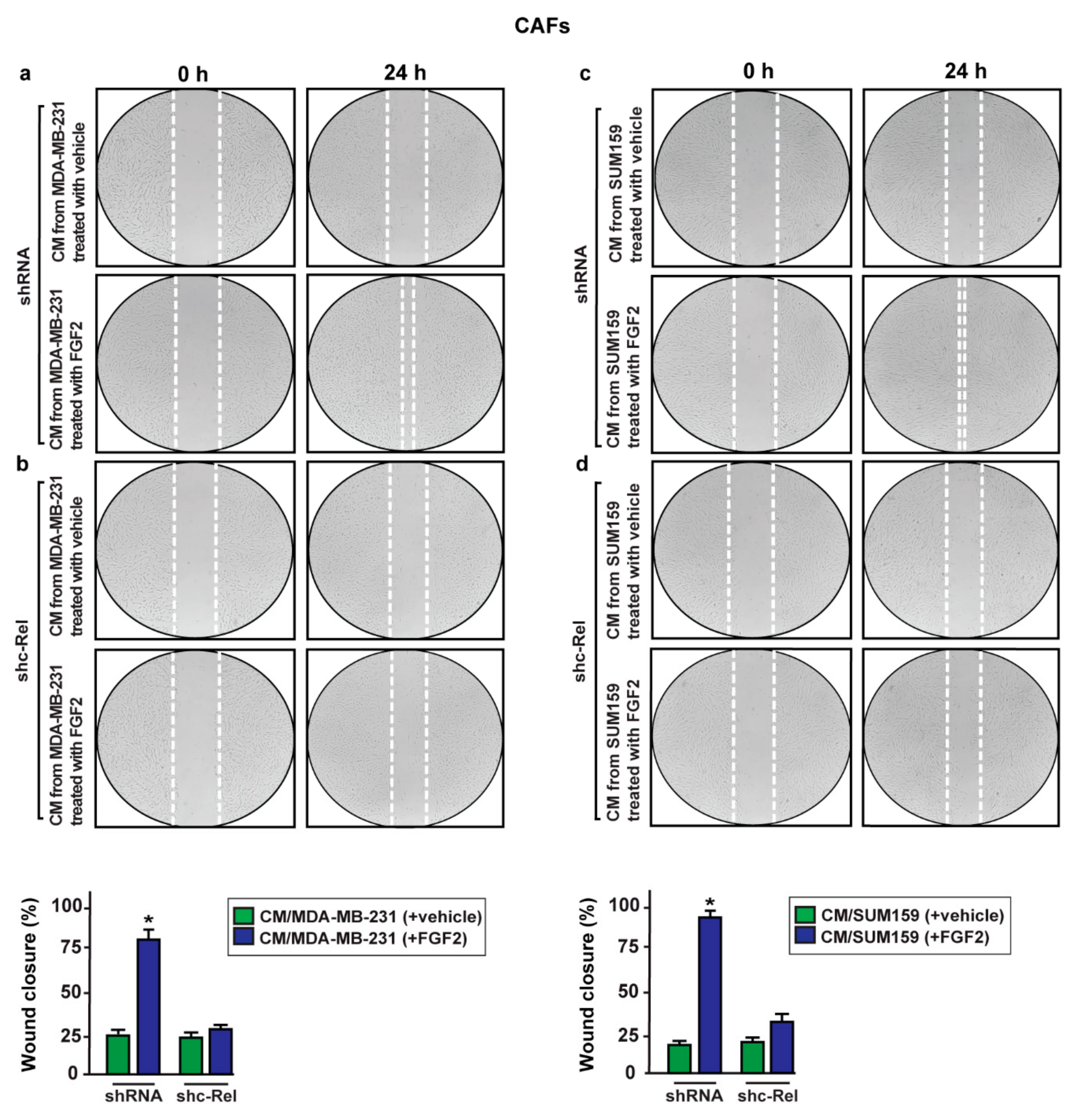
2.3. c-Rel Is Involved in the Upregulation of S100A4 Induced by FGF2/FGFR1 Signaling
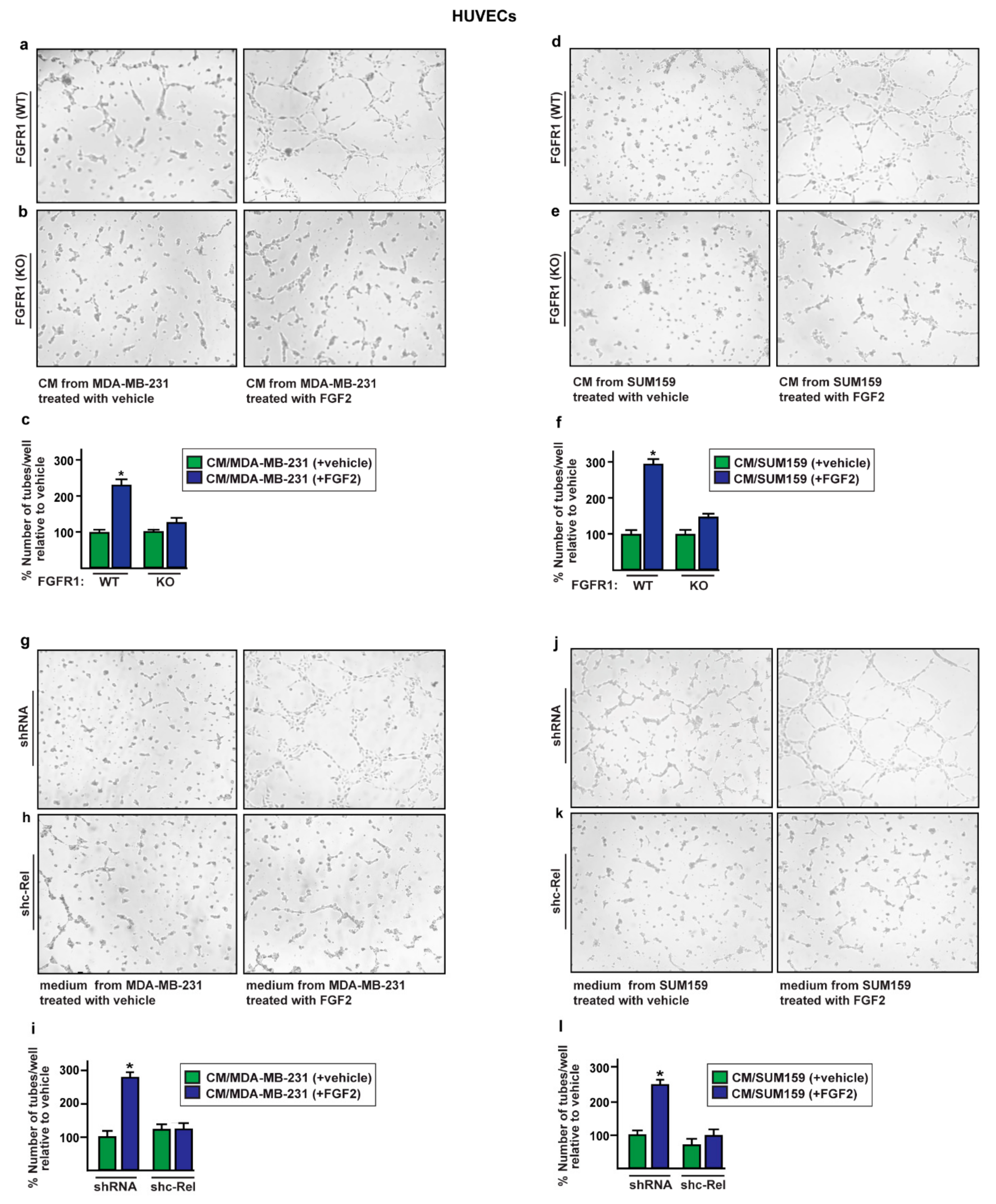
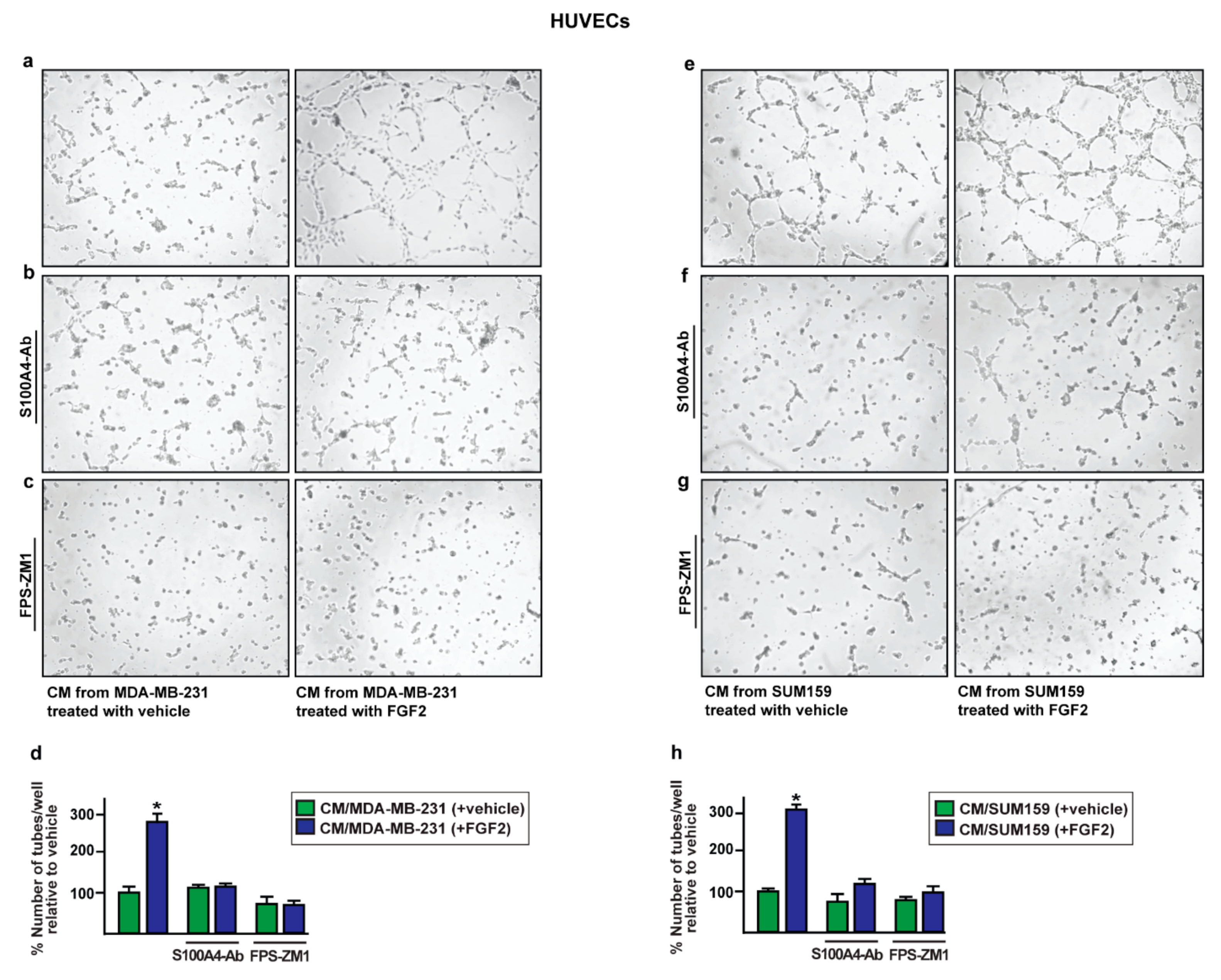
2.4. The Paracrine Activation of S100A4/RAGE Signaling Induces Endothelial Tube Formation in HUVECs
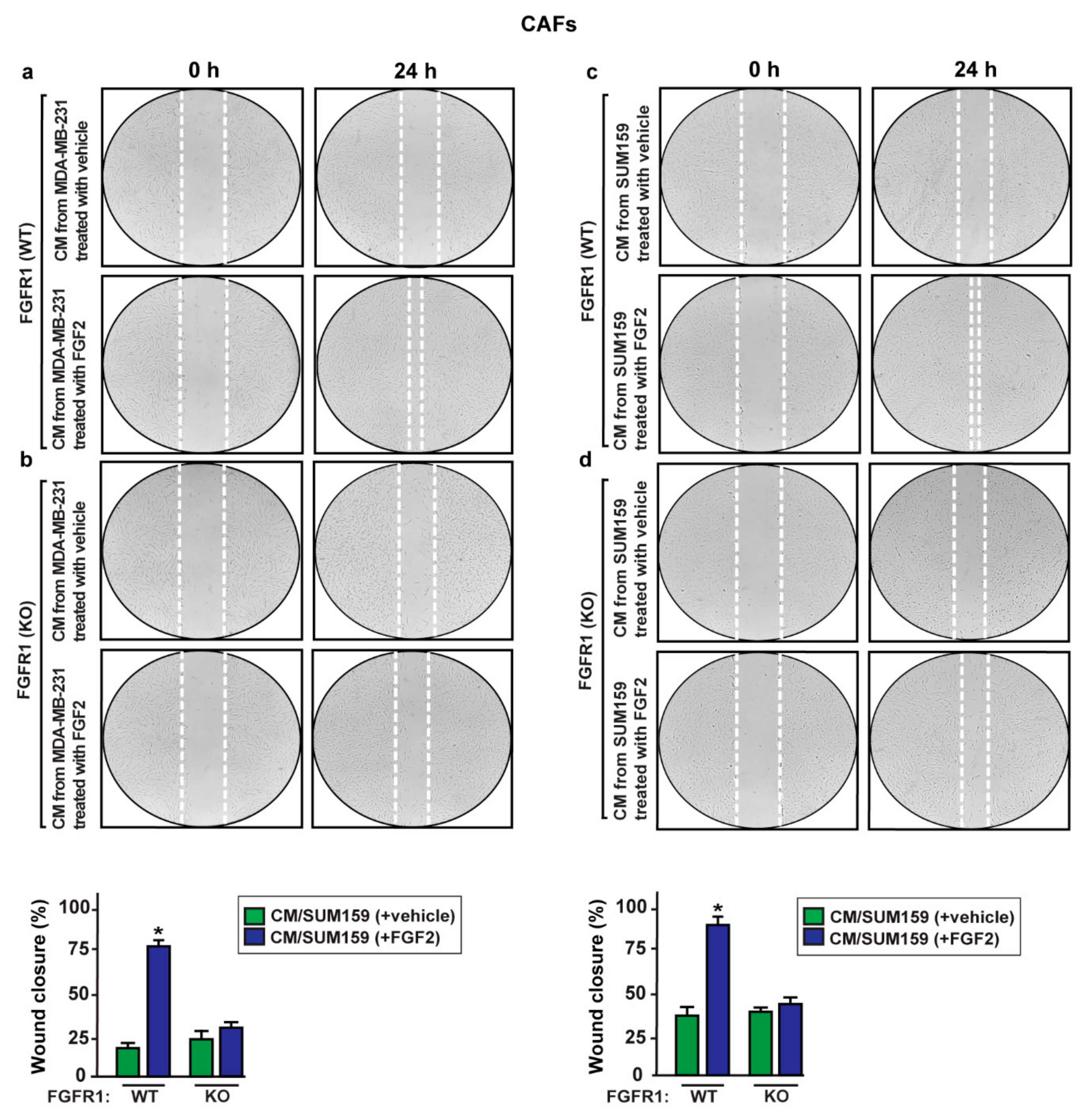
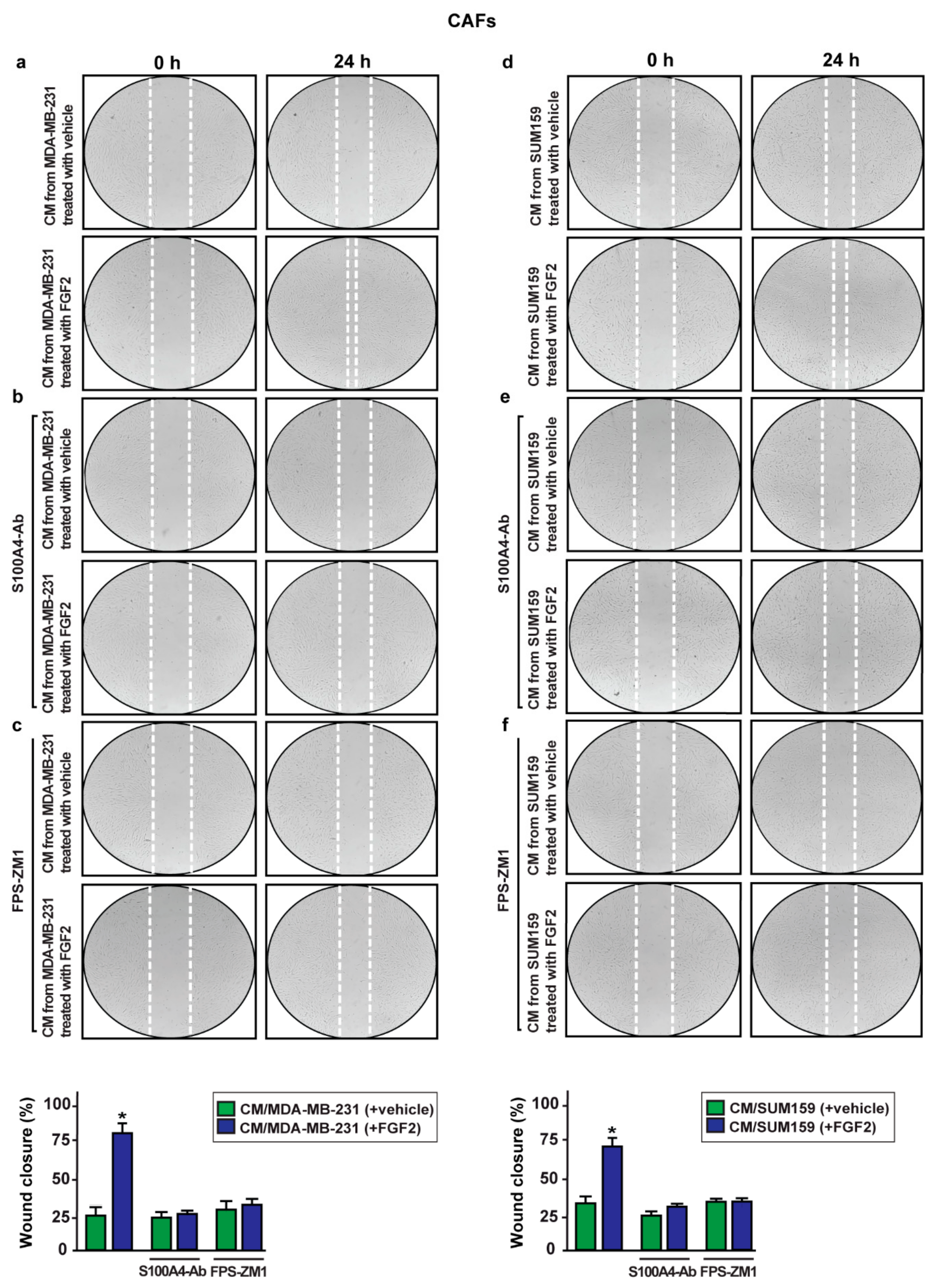
2.5. S100A4/RAGE Paracrine Activation Promotes Cell Migration in Breast Cancer-Associated Fibroblasts (CAFs)
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analyses
4.2. Reagents
4.3. Cell Cultures
4.4. Gene Expression Studies
4.5. Gene Silencing Experiments and Luciferase Assays
4.6. CRISPR/Cas9-Mediated FGFR1 Knockout
4.7. Western Blot Analysis
4.8. Immunofluorescence Microscopy
4.9. Chromatin Immunoprecipitation (ChIP) Assay
4.10. Conditioned Medium
4.11. Acetone Precipitation of Proteins
4.12. S100A4-Immunodepleted Conditioned Medium
4.13. Tube Formation Assay
4.14. Scratch Assay
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Sotiriou, C.; Pusztai, L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 2009, 360, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, P.S.; Parker, J.S.; Mullins, M.; Cheung, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.S.; Aksoy, B.A.B.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, S.-J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakman, C.; Viale, G.; Di Leo, A. Management of triple negative breast cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef]
- Venkitaraman, R. Triple-negative/basal-like breast cancer: Clinical, pathologic and molecular features. Expert Rev. Anticancer Ther. 2010, 10, 199–207. [Google Scholar] [CrossRef]
- Cheang, M.C.U.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [Green Version]
- Cocco, S.; Piezzo, M.; Calabrese, A.; Cianniello, D.; Caputo, R.; Di Lauro, V.; Fusco, G.; Di Gioia, G.; Licenziato, M.; de Laurentiis, M. Biomarkers in triple-negative breast cancer: State-of-the-art and future perspectives. Int. J. Mol. Sci. 2020, 21, 4579. [Google Scholar] [CrossRef]
- Cheng, C.L.; Thike, A.A.; Tan, S.Y.J.; Chua, P.J.; Bay, B.H.; Tan, P.H. Expression of FGFR1 is an independent prognostic factor in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 151, 99–111. [Google Scholar] [CrossRef]
- Sobhani, N.; Fan, C.; Flores-Villanueva, P.O.; Generali, D.; Li, Y. The Fibroblast Growth Factor Receptors in Breast Cancer: From Oncogenesis to Better Treatments. Int. J. Mol. Sci. 2020, 21, 2011. [Google Scholar] [CrossRef] [Green Version]
- Hui, Q.; Jin, Z.; Li, X.; Liu, C.; Wang, X. FGF family: From drug development to clinical application. Int. J. Mol. Sci. 2018, 19, 1875. [Google Scholar] [CrossRef] [Green Version]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Brady, N.J.; Chuntova, P.; Bade, L.K.; Schwertfeger, K.L. The FGF/FGF receptor axis as a therapeutic target in breast cancer. Expert Rev. Endocrinol. Metab. 2013, 8, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, N.E.; Dey, J.H. Potential for Targeting the Fibroblast Growth Factor Receptors in Breast Cancer. Cancer Res. 2010, 70, 5199–5202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275–5286. [Google Scholar] [CrossRef] [Green Version]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer HHS Public Access. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of Tumor Development and Metastasis Formation in Mice Lacking the S100A4 (mts1) Gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [Green Version]
- Grum-Schwensen, B.; Klingelhöfer, J.; Grigorian, M.; Almholt, K.; Nielsen, B.S.; Lukanidin, E.; Ambartsumian, N. Lung Metastasis Fails in MMTV-PyMT Oncomice Lacking S100A4 Due to a T-Cell Deficiency in Primary Tumors. Cancer Res. 2010, 70, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Grum-Schwensen, B.; Klingelhöfer, J.; Beck, M.; Bonefeld, M.M.; Hamerlik, P.; Guldberg, P.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. S100A4-neutralizing antibody suppresses spontaneous tumor progression, pre-metastatic niche formation and alters T-cell polarization balance. BMC Cancer 2015, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Hansen, B.; Örnås, D.; Grigorian, M.; Klingelhöfer, J.; Tulchinsky, E.; Lukanidin, E.; Ambartsumian, N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 2004, 23, 5487–5495. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Hansen, B.; Klingelhöfer, J.; Grum-Schwensen, B.; Christensen, A.; Andresen, S.; Kruse, C.; Hansen, T.; Ambartsumian, N.; Lukanidin, E.; Grigorian, M. Functional Significance of Metastasis-inducing S100A4(Mts1) in Tumor-Stroma Interplay. J. Biol. Chem. 2004, 279, 24498–24504. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Cabezón, T.; Celis, J.E.; Skibshøj, I.; Klingelhöfer, J.; Grigorian, M.; Gromov, P.; Rank, F.; Myklebust, J.H.; Mælandsmo, G.M.; Lukanidin, E.; et al. Expression of S100A4 by a variety of cell types present in the tumor microenvironment of human breast cancer. Int. J. Cancer 2007, 121, 1433–1444. [Google Scholar] [CrossRef]
- Ambartsumian, N.; Klingelhöfer, J.; Grigorian, M.; Christensen, C.; Kriajevska, M.; Tulchinsky, E.; Georgiev, G.; Berezin, V.; Bock, E.; Rygaard, J.; et al. The metastasis-associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene 2001, 20, 4685–4695. [Google Scholar] [CrossRef] [Green Version]
- Boye, K.; Mælandsmo, G.M. S100A4 and Metastasis. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef]
- Siddique, H.R.; Adhami, V.M.; Parray, A.; Johnson, J.J.; Siddiqui, I.A.; Shekhani, M.T.; Murtaza, I.; Ambartsumian, N.; Konety, B.R.; Mukhtar, H.; et al. The S100A4 Oncoprotein Promotes Prostate Tumorigenesis in a Transgenic Mouse Model: Regulating NFκB through the RAGE Receptor. Genes Cancer 2013, 4, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, J.L.; Padilla, L.; Dakhel, S.; Coll, T.; Hervas, R.; Adan, J.; Masa, M.; Mitjans, F.; Martinez, J.M.; Coma, S.; et al. Therapeutic Targeting of Tumor Growth and Angiogenesis with a Novel Anti-S100A4 Monoclonal Antibody. PLoS ONE 2013, 8, e72480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, N.; Belter, B.; Pietzsch, J. Extracellular S100A4 affects endothelial cell integrity and stimulates transmigration of A375 melanoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nankali, M.; Karimi, J.; Goodarzi, M.T.; Saidijam, M.; Khodadadi, I.; Razavi, A.N.E.; Rahimi, F. Increased Expression of the Receptor for Advanced Glycation End-Products (RAGE) Is Associated with Advanced Breast Cancer Stage. Oncol. Res. Treat. 2016, 39, 622–628. [Google Scholar] [CrossRef]
- Nasser, M.W.; Wani, N.A.; Ahirwar, D.K.; Powell, C.A.; Ravi, J.; Elbaz, M.; Zhao, H.; Padilla, L.; Zhang, X.; Shilo, K.; et al. RAGE Mediates S100A7-Induced Breast Cancer Growth and Metastasis by Modulating the Tumor Microenvironment. Cancer Res. 2015, 75, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- El-Far, A.H.; Sroga, G.; Al Jaouni, S.K.; Mousa, S.A. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 3613. [Google Scholar] [CrossRef]
- Chen, P.S.; Wang, M.Y.; Wu, S.N.; Su, J.L.; Hong, C.C.; Chuang, S.E.; Chen, M.W.; Hua, K.T.; Wu, Y.L.; Cha, S.T.; et al. CTGF enhances the motility of breast cancer cells via an integrin-αvβ3-ERK1/2-dependent S100A4-upregulated pathway. J. Cell Sci. 2007, 120, 2053–2065. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, A.; Hayashi, T.; Kikuchi, N.; Hayashi, A.; Fuseya, C.; Shiozawa, T.; Konishi, I. Hypoxia upregulates ovarian cancer invasiveness via the binding of HIF-1α to a hypoxia-induced, methylation-free hypoxia response element of S100A4 gene. Int. J. Cancer 2012, 131, 1755–1767. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.G.; Meng, Q.; Qi, F.M.; Yang, Q.F. Blocking TGF-β inhibits breast cancer cell invasiveness via ERK/S100A4 signal. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3844–3853. [Google Scholar]
- Hellinger, J.W.; Hüchel, S.; Goetz, L.; Bauerschmitz, G.; Emons, G.; Gründker, C. Inhibition of cyr61-s100a4 axis limits breast cancer invasion. Front. Oncol. 2019, 9, 1074. [Google Scholar] [CrossRef]
- Forst, B.; Hansen, M.T.; Klingelhöfer, J.; Möller, H.D.; Nielsen, G.H.; Grum-Schwensen, B.; Ambartsumian, N.; Lukanidin, E.; Grigorian, M. Metastasis-inducing S100A4 and RANTES cooperate in promoting tumor progression in mice. PLoS ONE 2010, 5, e10374. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Liang, Y.; Diao, X.; Chen, Q. S100A4 promotes invasion and angiogenesis in breast cancer MDA-MB-231 cells by upregulating matrix metalloproteinase-13. Acta Biochim. Pol. 2012, 59, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Egeland, E.V.; Boye, K.; Park, D.; Synnestvedt, M.; Sauer, T.; Sauer, T.; Geisler, J.; Hofvind, S.; Bathen, T.F.; Borgen, E.; et al. Prognostic significance of S100A4-expression and subcellular localization in early-stage breast cancer. Breast Cancer Res. Treat. 2017, 162, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Su, W.C.; Lin, P.W.; Guo, H.R.; Chang, T.W.; Chen, H.H.W. Expression of S100A4 and Met: Potential Predictors for Metastasis and Survival in Early-Stage Breast Cancer. Oncology 2004, 66, 429–438. [Google Scholar] [CrossRef]
- Rudland, P.S.; Platt-Higgins, A.; Renshaw, C.; West, C.R.; Winstanley, J.H.R.; Robertson, L.; Barraclough, R. Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res. 2000, 60, 1595–1603. [Google Scholar]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef]
- Lee, J.G.; Kay, E.D.P. NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1530–1538. [Google Scholar] [CrossRef]
- Wang, C.; Ke, Y.; Liu, S.; Pan, S.; Liu, Z.; Zhang, H.; Fan, Z.; Zhou, C.; Liu, J.; Wang, F. Ectopic fibroblast growth factor receptor 1 promotes inflammation by promoting nuclear factor-кB signaling in prostate cancer cells. J. Biol. Chem. 2018, 293, 14839–14849. [Google Scholar] [CrossRef] [Green Version]
- Vandermoere, F.; El Yazidi-Belkoura, I.; Adriaenssens, E.; Lemoine, J.; Hondermarck, H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-κB activation induced via interaction between Akt and IκB kinase-β in breast cancer cells. Oncogene 2005, 24, 5482–5491. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-S.; Min, K.-S.; Jeong, D.-H.; Jang, J.-H.; Kim, H.-W.; Kim, E.-C. Effects of Fibroblast Growth Factor-2 on the Expression and Regulation of Chemokines in Human Dental Pulp Cells. J. Endod. 2010, 36, 1824–1830. [Google Scholar] [CrossRef]
- Santolla, M.F.; Lappano, R.; Cirillo, F.; Rigiracciolo, D.C.; Sebastiani, A.; Abonante, S.; Tassone, P.; Tagliaferri, P.; Di Martino, M.T.; Maggiolini, M.; et al. miR-221 stimulates breast cancer cells and cancer-associated fibroblasts (CAFs) through selective interference with the A20/c-Rel/CTGF signaling. J. Exp. Clin. Cancer Res. 2018, 37, 94. [Google Scholar] [CrossRef]
- Belguise, K.; Sonenshein, G.E. PKCθ promotes c-Rel-driven mammary tumorigenesis in mice and humans by repressing estrogen receptor α synthesis. J. Clin. Investig. 2007, 117, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Romieu-Mourez, R.; Kim, D.W.; Shin, S.M.; Demicco, E.G.; Landesman-Bollag, E.; Seldin, D.C.; Cardiff, R.D.; Sonenshein, G.E. Mouse Mammary Tumor Virus c-rel Transgenic Mice Develop Mammary Tumors. Mol. Cell. Biol. 2003, 23, 5738–5754. [Google Scholar] [CrossRef] [Green Version]
- Ochiya, T.; Takenaga, K.; Endo, H. Silencing of S100A4, a metastasis-associated protein, in endothelial cells inhibits tumor angiogenesis and growth. Angiogenesis 2014, 17, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Donato, R. RAGE: A single receptor for several ligands and different cellular responses: The case of certain S100 proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef]
- Shubbar, E.; Vegfors, J.; Carlström, M.; Petersson, S.; Enerbäck, C. Psoriasin (S100A7) increases the expression of ROS and VEGF and acts through RAGE to promote endothelial cell proliferation. Breast Cancer Res. Treat. 2012, 134, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Laplagne, C.; Domagala, M.; Le Naour, A.; Quemerais, C.; Hamel, D.; Fournié, J.J.; Couderc, B.; Bousquet, C.; Ferrand, A.; Poupot, M. Latest advances in targeting the tumor microenvironment for tumor suppression. Int. J. Mol. Sci. 2019, 20, 4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Mizejewski, G.J. Breast cancer, metastasis, and the microenvironment: Disabling the tumor cell-to-stroma communication network. J. Cancer Metastasis Treat. 2019, 5, 35. [Google Scholar] [CrossRef]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the cancer microenvironment as a target for therapeutic interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef]
- Akl, M.R.; Nagpal, P.; Ayoub, N.M.; Tai, B.; Prabhu, S.A.; Capac, C.M.; Gliksman, M.; Goy, A.; Suh, K.S. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016, 7, 44735–44762. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; Van Diest, P.J.; De Jong, J.S.; Van Der Groep, P.; Van Der Valk, P.; Van Der Wall, E. Hypoxia-inducible factor-1α is associated with angiogenesis, and expression of bFGF, PDGF-BB, and EGFR in invasive breast cancer. Histopathology 2005, 46, 31–36. [Google Scholar] [CrossRef]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells Toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.; Kim, D.; Lee, Y.; Jang, J.; Surh, Y. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinog. 2020, 59, 1028–1040. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Ebralidze, A.; Tulchinsky, E.; Grigorian, M.; Afanasyeva, A.; Senin, V.; Revazova, E.; Lukanidin, E. Isolation and characterization of a gene specifically expressed in different metastatic cells and whose deduced gene product has a high degree of homology to a Ca2+-binding protein family. Genes Dev. 1989, 3, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Garrett, S.C.; Varney, K.M.; Weber, D.J.; Bresnick, A.R. S100A4, a mediator of metastasis. J. Biol. Chem. 2006, 281, 677–680. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Qu, J.; Li, C.; Wang, X.; Li, Y.; Zhang, S. Role of metastasis-induced protein S100A4 in human non-tumor pathophysiologies. Cell Biosci. 2017, 7, 64. [Google Scholar] [CrossRef]
- Leśniak, W. Epigenetic regulation of S100 protein expression. Clin. Epigenetics 2011, 2, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Day, T.K.; Bianco-Miotto, T. Common gene pathways and families altered by DNA methylation in breast and prostate cancers. Endocr. Relat. Cancer 2013, 20, R215–R232. [Google Scholar] [CrossRef] [Green Version]
- Ambartsumian, N.; Klingelhöfer, J.; Grigorian, M. The multifaceted S100A4 protein in cancer and inflammation. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1929, pp. 339–365. [Google Scholar] [CrossRef]
- Cortés Sempere, M.; Rodríguez Fanjul, V.; Sánchez Pérez, I.; Perona, R. The role of the NFkappaB signalling pathway in cancer. Clin. Transl. Oncol. 2008, 10, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.; Mori, H.; Fata, J.; Bascom, J.; Øyjord, T.; Mælandsmo, G.M.; Bissell, M. The metastasis-promoting protein S100A4 regulates mammary branching morphogenesis. Dev. Biol. 2011, 352, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Hiruta, A.; Oguri, Y.; Yokoi, A.; Matsumoto, T.; Oda, Y.; Tomohiro, M.; Hashimura, M.; Jiang, Z.; Tochimoto, M.; Nakagawa, M.; et al. S100A4/Nonmuscle Myosin IIA/p53 Axis Contributes to Aggressive Features in Ovarian High-Grade Serous Carcinoma. Am. J. Pathol. 2020, 190, 2304–2316. [Google Scholar] [CrossRef]
- Chen, H.; Xu, C.; Jin, Q.; Liu, Z. S100 protein family in human cancer. Am. J. Cancer Res. 2014, 4, 89–115. [Google Scholar] [PubMed]
- Kriajevska, M.; Fischer-Larsen, M.; Moertz, E.; Vorm, O.; Tulchinsky, E.; Grigorian, M.; Ambartsumian, N.; Lukanidin, E. Liprin β1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J. Biol. Chem. 2002, 277, 5229–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orre, L.M.; Panizza, E.; Kaminskyy, V.O.; Vernet, E.; Gräslund, T.; Zhivotovsky, B.; Lehtiö, J. S100A4 interacts with p53 in the nucleus and promotes p53 degradation. Oncogene 2013, 32, 5531–5540. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Elbaz, M.; Ahirwar, D.K.; Ganju, R.K. Conditioning solid tumor microenvironment through inflammatory chemokines and S100 family proteins. Cancer Lett. 2015, 365, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE mediates the pro-migratory response of extracellular S100A4 in human thyroid cancer cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4 + stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Santolla, M.F.; De Francesco, E.M.; Lappano, R.; Rosano, C.; Abonante, S.; Maggiolini, M. Niacin activates the G protein estrogen receptor (GPER)-mediated signalling. Cell. Signal. 2014, 26, 1466–1475. [Google Scholar] [CrossRef]
- Khaled, W.T.; Lee, S.C.; Stingl, J.; Chen, X.; Ali, H.R.; Rueda, O.M.; Hadi, F.; Wang, J.; Yu, Y.; Chin, S.F.; et al. BCL11A is a Triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Sastry, S.K.; O’Connor, K.L. Src kinase pathway is involved in NFAT5-mediated S100A4 induction by hyperosmotic stress in colon cancer cells. Am. J. Physiol. Cell Physiol. 2011, 300, C1155–C1163. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.P.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Santolla, M.F.; Avino, S.; Pellegrino, M.; De Francesco, E.M.; De Marco, P.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Rigiracciolo, D.C.; Scarpelli, A.; et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis. 2015, 6, e1834. [Google Scholar] [CrossRef]
- Zhang, Y.; Bottinelli, D.; Lisacek, F.; Luban, J.; Strambio-De-Castillia, C.; Varesio, E.; Hopfgartner, G. Optimization of human dendritic cell sample preparation for mass spectrometry-based proteomic studies. Anal. Biochem. 2015, 484, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Fic, E.; Kedracka-Krok, S.; Jankowska, U.; Pirog, A.; Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis 2010, 31, 3573–3579. [Google Scholar] [CrossRef]
- Gelsomino, L.; Giordano, C.; La Camera, G.; Sisci, D.; Marsico, S.; Campana, A.; Tarallo, R.; Rinaldi, A.; Fuqua, S.; Leggio, A.; et al. Leptin signaling contributes to aromatase inhibitor resistant breast cancer cell growth and activation of macrophages. Biomolecules 2020, 10, 543. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Catalano, S.; Gelsomino, L.; Marsico, S.; Giordano, C.; Panza, S.; Bonofiglio, D.; Bossi, G.; Covington, K.R.; Fuqua, S.A.W.; et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 2012, 72, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Patil, P.U.; D’Ambrosio, J.; Inge, L.J.; Mason, R.W.; Rajasekaran, A.K. Carcinoma cells induce lumen filling and EMT in epithelial cells through soluble E-cadherin-mediated activation of EGFR. J. Cell Sci. 2015, 128, 4366–4379. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santolla, M.F.; Talia, M.; Maggiolini, M. S100A4 Is Involved in Stimulatory Effects Elicited by the FGF2/FGFR1 Signaling Pathway in Triple-Negative Breast Cancer (TNBC) Cells. Int. J. Mol. Sci. 2021, 22, 4720. https://doi.org/10.3390/ijms22094720
Santolla MF, Talia M, Maggiolini M. S100A4 Is Involved in Stimulatory Effects Elicited by the FGF2/FGFR1 Signaling Pathway in Triple-Negative Breast Cancer (TNBC) Cells. International Journal of Molecular Sciences. 2021; 22(9):4720. https://doi.org/10.3390/ijms22094720
Chicago/Turabian StyleSantolla, Maria Francesca, Marianna Talia, and Marcello Maggiolini. 2021. "S100A4 Is Involved in Stimulatory Effects Elicited by the FGF2/FGFR1 Signaling Pathway in Triple-Negative Breast Cancer (TNBC) Cells" International Journal of Molecular Sciences 22, no. 9: 4720. https://doi.org/10.3390/ijms22094720
APA StyleSantolla, M. F., Talia, M., & Maggiolini, M. (2021). S100A4 Is Involved in Stimulatory Effects Elicited by the FGF2/FGFR1 Signaling Pathway in Triple-Negative Breast Cancer (TNBC) Cells. International Journal of Molecular Sciences, 22(9), 4720. https://doi.org/10.3390/ijms22094720