
Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Tumors with Follicular Differentiation
2.1. Malignant Tumors with Follicular Differentiation
2.1.1. Pilomatrical Carcinoma
2.1.2. Proliferating Trichilemmal Tumor
2.1.3. Trichoblastic Carcinoma/Carcinosarcoma
2.1.4. Tricholemmal Carcinoma
2.2. Bening Tumors with Follicular Differentiation
2.2.1. Trichoblastoma
2.2.2. Pilomatricoma
2.2.3. Trichilemmoma
2.2.4. Trichofolliculoma
2.2.5. Pilar Sheath Acanthoma
2.2.6. Tumor of the Follicular Infundibulum
2.2.7. Melanocytic Matricoma
2.2.8. Spindle Cell-Predominant Trichodiscoma
3. Conclusions

{kind=link}
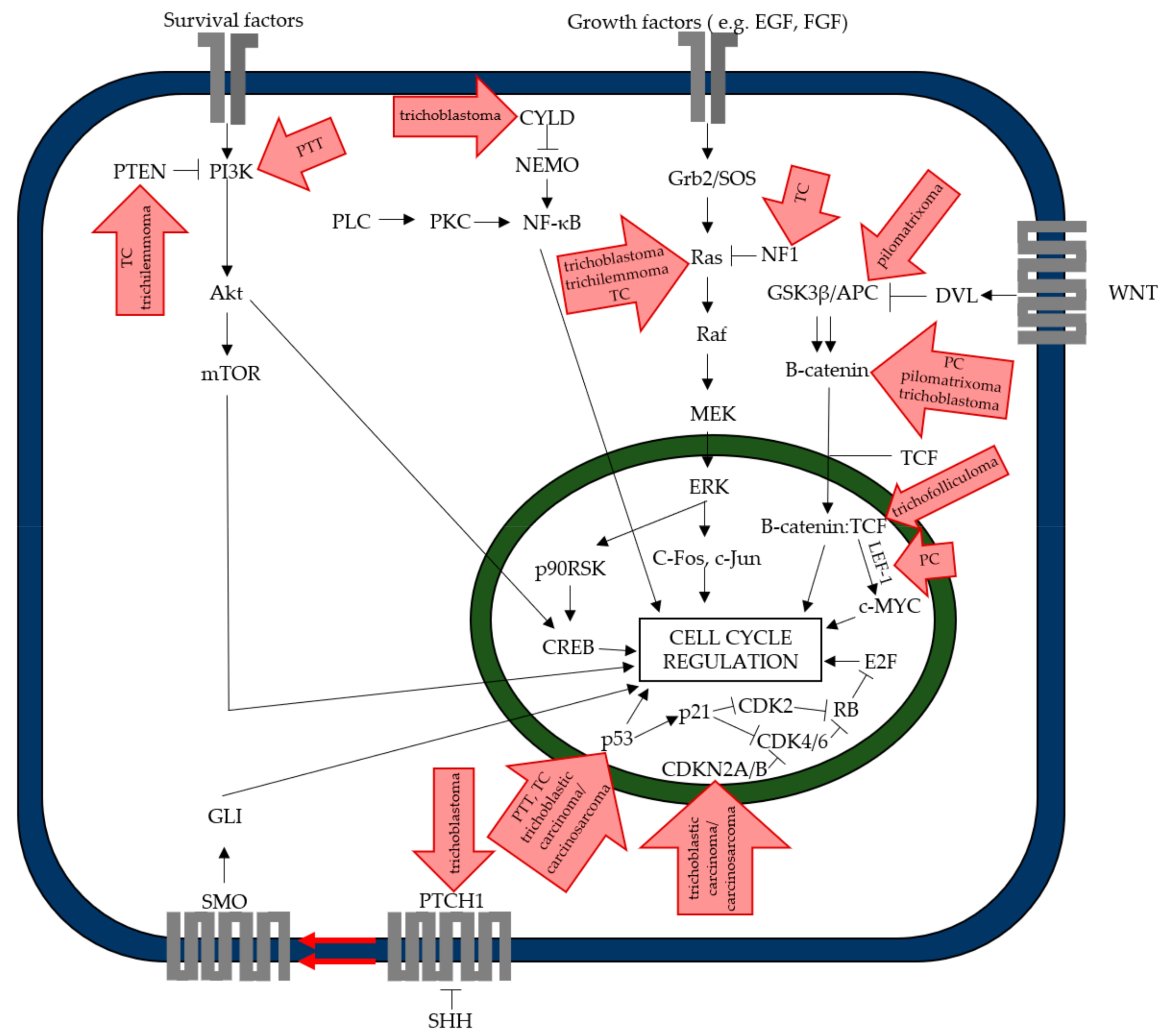{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Surgery | Chemotherapy (Agents) | Reference | Radiotherapy | Reference |
|---|---|---|---|---|---|
| Pilomatrical carcinoma | R | NR | [25,29,72,104] | A or NA: 45 Gy, 4.5 Gy/fraction; A after LND | [25,29,30,31,32,72,104,105] |
| Proliferating trichilemmal tumor | R | MD: cisplatin, doxorubicin, vinca alkaloids, 5-fluorouracil PI3K inhibitor | [12,41,43,106] | A, MD | [12,39,42,43,61,106] |
| Trichoblastic carcinoma/carcinosarcoma | R | pharmorubicin-cisplatin, vismodegib, sunitinib | [45,46,51,52,107] | LD | [45,46,107,108,109] |
| Tricholemmal carcinoma | R | LD | [54,59,63,64,65,110] | LD | [54,62,63] |
| Trichoblastoma | R | ND | ND | ||
| Pilomatrixoma | R | ND | ND | ||
| Trichilemmoma | R | ND | ND | ||
| Trichofolliculoma | R | ND | ND | ||
| Pilar sheath acanthoma | R | ND | ND | ||
| Tumor of the follicular infundibulum | R | ND | ND | ||
| Melanocytic matricoma | R | ND | ND | ||
| Spindle cell-predominant trichodiscoma | R | ND | ND |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elder, D.E.; Massi, D.; Scolyer, R.A.; Willemze, R. WHO Classification of Skin Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2018; pp. 195–210. [Google Scholar]
- Tellechea, O.; Cardoso, J.C.; Reis, J.P.; Ramos, L.; Gameiro, A.R.; Coutinho, I.; Baptista, A.P. Benign follicular tumors. Bras. Derm. 2015, 90, 780–796; quiz 797–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashinoff, R.; Jacobson, M.; Belsito, D.V. Rombo syndrome: A second case report and review. J. Am. Acad. Dermatol. 1993, 28, 1011–1014. [Google Scholar] [CrossRef]
- Kazakov, D.V. Brooke-Spiegler Syndrome and Phenotypic Variants: An Update. Head Neck Pathol. 2016, 10, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Walker, L.J.; Bowling, J.; Audisio, R.A. Cowden syndrome. Cancer Treat. Rev. 2010, 36, 577–583. [Google Scholar] [CrossRef]
- Stevenson, P.; Rodins, K.; Susman, R. The association between multiple pilomatrixomas and APC gene mutations. Australas J. Derm. 2018, 59, e273–e274. [Google Scholar] [CrossRef]
- Trufant, J.; Kurz, W.; Frankel, A.; Muthusamy, V.; McKinnon, W.; Greenblatt, M.; Lazar, A.; Cook, D.; Bosenberg, M. Familial multiple pilomatrixomas as a presentation of attenuated adenomatosis polyposis coli. J. Cutan. Pathol. 2012, 39, 440–443. [Google Scholar] [CrossRef]
- Rohwedder, A.; Keminer, O.; Hendricks, C.; Schaller, J. Detection of HPV DNA in trichilemmomas by polymerase chain reaction. J. Med. Virol. 1997, 51, 119–125. [Google Scholar] [CrossRef]
- Tumminello, K.; Hosler, G.A. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J. Cutan. Pathol. 2018, 45, 318–324. [Google Scholar] [CrossRef]
- Kazakov, D.V.; Sima, R.; Vanecek, T.; Kutzner, H.; Palmedo, G.; Kacerovska, D.; Grossmann, P.; Michal, M. Mutations in exon 3 of the CTNNB1 gene (beta-catenin gene) in cutaneous adnexal tumors. Am. J. Derm. 2009, 31, 248–255. [Google Scholar] [CrossRef]
- Takata, M.; Rehman, I.; Rees, J.L. A trichilemmal carcinoma arising from a proliferating trichilemmal cyst: The loss of the wild-type p53 is a critical event in malignant transformation. Hum. Pathol. 1998, 29, 193–195. [Google Scholar] [CrossRef]
- Gallant, J.N.; Sewell, A.; Almodovar, K.; Wang, Q.; Dahlman, K.B.; Abramson, R.G.; Kapp, M.E.; Brown, B.T.; Boyd, K.L.; Gilbert, J.; et al. Genomic landscape of a metastatic malignant proliferating tricholemmal tumor and its response to PI3K inhibition. NPJ Precis. Oncol. 2019, 3, 5. [Google Scholar] [CrossRef]
- Nakai, N.; Takenaka, H.; Hamada, S.; Kishimoto, S. Identical p53 gene mutation in malignant proliferating trichilemmal tumour of the scalp and small cell carcinoma of the common bile duct: The necessity for therapeutic caution? Br. J. Derm. 2008, 159, 482–485. [Google Scholar] [CrossRef]
- Ha, J.H.; Lee, C.; Lee, K.S.; Pak, C.S.; Sun, C.H.; Koh, Y.; Chang, H. The molecular pathogenesis of Trichilemmal carcinoma. BMC Cancer 2020, 20, 516. [Google Scholar] [CrossRef]
- Shen, A.S.; Peterhof, E.; Kind, P.; Rutten, A.; Zelger, B.; Landthaler, M.; Berneburg, M.; Hafner, C.; Groesser, L. Activating mutations in the RAS/mitogen-activated protein kinase signaling pathway in sporadic trichoblastoma and syringocystadenoma papilliferum. Hum. Pathol. 2015, 46, 272–276. [Google Scholar] [CrossRef]
- Hafner, C.; Schmiemann, V.; Ruetten, A.; Coras, B.; Landthaler, M.; Reifenberger, J.; Vogt, T. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum. Pathol. 2007, 38, 1496–1500. [Google Scholar] [CrossRef]
- Lazar, A.J.; Calonje, E.; Grayson, W.; Dei Tos, A.P.; Mihm, M.C., Jr.; Redston, M.; McKee, P.H. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J. Cutan. Pathol. 2005, 32, 148–157. [Google Scholar] [CrossRef]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Hassanein, A.M.; Glanz, S.M. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br. J. Derm. 2004, 150, 511–516. [Google Scholar] [CrossRef]
- Urabe, K.; Xia, J.; Masuda, T.; Moroi, Y.; Furue, M.; Matsumoto, T. Pilomatricoma-like changes in the epidermoid cysts of Gardner syndrome with an APC gene mutation. J. Derm. 2004, 31, 255–257. [Google Scholar] [CrossRef]
- Agoston, A.T.; Liang, C.W.; Richkind, K.E.; Fletcher, J.A.; Vargas, S.O. Trisomy 18 is a consistent cytogenetic feature in pilomatricoma. Mod. Pathol. 2010, 23, 1147–1150. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Huang, W.C.; Jhuang, J.Y.; Jeng, Y.M.; Cheng, M.L.; Chiu, H.Y.; Kuo, K.T.; Liau, J.Y. Frequent activating HRAS mutations in trichilemmoma. Br. J. Derm. 2014, 171, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Watanabe, K.; Fallahi, M.; Lee, B.; Afetian, M.E.; Rheaume, C.; Wu, D.; Horsley, V.; Dai, X. Pygo2 regulates beta-catenin-induced activation of hair follicle stem/progenitor cells and skin hyperplasia. Proc. Natl. Acad. Sci. USA 2014, 111, 10215–10220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremnes, R.M.; Kvamme, J.M.; Stalsberg, H.; Jacobsen, E.A. Pilomatrix carcinoma with multiple metastases: Report of a case and review of the literature. Eur. J. Cancer 1999, 35, 433–437. [Google Scholar] [CrossRef]
- Herrmann, J.L.; Allan, A.; Trapp, K.M.; Morgan, M.B. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J. Am. Acad. Derm. 2014, 71, 38–43. [Google Scholar] [CrossRef]
- Nogal, P.; Bartkowiak, E.; Iwanik, K.; Wierzbicka, M. Common sense and tumor treatment. A case of pilomatrical carcinoma in a 21-year-old patient with surprisingly rapid tumor progression. Oral Oncol. 2020, 112, 105007. [Google Scholar] [CrossRef]
- Sable, D.; Snow, S.N. Pilomatrix carcinoma of the back treated by mohs micrographic surgery. Derm. Surg. 2004, 30, 1174–1176. [Google Scholar] [CrossRef]
- Papadakis, M.; de Bree, E.; Floros, N.; Giannikaki, E.; Xekalou, A.; Manios, A. Pilomatrix carcinoma: More malignant biological behavior than was considered in the past. Mol. Clin. Oncol. 2017, 6, 415–418. [Google Scholar] [CrossRef]
- Tselis, N.; Heyd, R.; Vogt, H.G.; Zamboglou, N. Pilomatrix carcinoma with lymph node and pulmonary metastases. Strahlenther Onkol. 2006, 182, 727–732. [Google Scholar] [CrossRef]
- Sau, P.; Lupton, G.P.; Graham, J.H. Pilomatrix carcinoma. Cancer 1993, 71, 2491–2498. [Google Scholar] [CrossRef]
- Gould, E.; Kurzon, R.; Kowalczyk, A.P.; Saldana, M. Pilomatrix carcinoma with pulmonary metastasis. Report of a case. Cancer 1984, 54, 370–372. [Google Scholar] [CrossRef]
- Waqas, O.; Faisal, M.; Haider, I.; Amjad, A.; Jamshed, A.; Hussain, R. Retrospective study of rare cutaneous malignant adnexal tumors of the head and neck in a tertiary care cancer hospital: A case series. J. Med. Case Rep. 2017, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.W. Proliferating Epidermoid Cysts. Arch. Dermatol. 1966, 94, 11–19. [Google Scholar] [CrossRef]
- ElBenaye, J.; Sinaa, M.; Elkhachine, Y.; Sakkah, A.; Jakar, A.; Elhaouri, M. Proliferating trichilemmal cyst of the scalp: Nine cases and literature review. Otorhinolaryngol. Head Neck Surg. 2018, 3. [Google Scholar] [CrossRef]
- Satyaprakash, A.K.; Sheehan, D.J.; Sangueza, O.P. Proliferating trichilemmal tumors: A review of the literature. Derm. Surg 2007, 33, 1102–1108. [Google Scholar] [CrossRef]
- Alici, O.; Keles, M.K.; Kurt, A. A Rare Cutaneous Adnexal Tumor: Malignant Proliferating Trichilemmal Tumor. Case Rep. Med. 2015, 2015, 742920. [Google Scholar] [CrossRef] [Green Version]
- Paolino, G.; Pampena, R.; Grassi, S.; Mercuri, S.R.; Cardone, M.; Corsetti, P.; Moliterni, E.; Muscianese, M.; Rossi, A.; Frascione, P.; et al. Alopecia neoplastica as a sign of visceral malignancies: A systematic review. J. Eur. Acad. Derm. Venereol. 2019, 33, 1020–1028. [Google Scholar] [CrossRef]
- Adegun, O.K.; Morley, S.; Kalavrezos, N.; Jay, A. Proliferating trichilemmal tumour: Diagnostic challenge on core biopsy. BMJ Case Rep. 2019, 12. [Google Scholar] [CrossRef]
- Ye, J.; Nappi, O.; Swanson, P.E.; Patterson, J.W.; Wick, M.R. Proliferating Pilar Tumors. Am. J. Clin. Pathol. 2004, 122, 566–574. [Google Scholar] [CrossRef]
- Fieleke, D.R.; Goldstein, G.D. Malignant proliferating trichilemmal tumor treated with Mohs surgery: Proposed protocol for diagnostic work-up and treatment. Derm. Surg. 2015, 41, 292–294. [Google Scholar] [CrossRef]
- Hayashi, I.; Harada, T.; Muraoka, M.; Ishii, M. Malignant proliferating trichilemmal tumour and CAV (cisplatin, adriamycin, vindesine) treatment. Br. J. Derm. 2004, 150, 156–157. [Google Scholar] [CrossRef]
- Dubhashi, S.P.; Jadhav, S.K.; Parasnis, A.; Patil, C.S. Recurrent malignant proliferating trichilemmal tumor with lymph node metastasis in a young woman. J. Postgrad. Med. 2014, 60, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.; Roth, K.; Yu, E. Malignant Proliferating Trichilemmal Tumor Treated with Radical Radiotherapy: A Case Report and Literature Review. Cureus 2017, 9, e999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.A.; Muller, S.; Chaudahri, P.J.; Carlson, J.A. Cutaneous carcinosarcoma: Adnexal vs. epidermal types define high- and low-risk tumors. Results of a meta-analysis. J. Cutan. Pathol. 2005, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Brown, A.; Osipov, V. Trichoblastic carcinosarcoma in a 34-year-old woman with histopathologic and molecular analysis, including re-demonstration of a CDKN2A p.(R58*) mutation. J. Cutan. Pathol. 2021, 48, 334–339. [Google Scholar] [CrossRef]
- Regauer, S.; Beham-Schmid, C.; Okcu, M.; Hartner, E.; Mannweiler, S. Trichoblastic carcinoma (“malignant trichoblastoma”) with lymphatic and hematogenous metastases. Mod. Pathol. 2000, 13, 673–678. [Google Scholar] [CrossRef]
- Nair, I.M.; Gharpuray-Pandit, D.; Cardozo, C. A case of trichoblastic carcinosarcoma with review of literature. Hum. Pathol. Case Rep. 2021, 24. [Google Scholar] [CrossRef]
- Yaacoub, E.; El Borgi, J.; Challita, R.; Sleiman, Z.; Ghanime, G. Pinna high grade trichoblastic carcinoma, a report. Clin. Pr. 2020, 10, 1204. [Google Scholar] [CrossRef]
- Li, J.J.X.; Ng, J.K.M.; Choi, P.C.L.; Lee, J.H.S.; Yu, M.Y. Trichoblastic Carcinosarcoma Arising From the Vagina: A Case Report With Comprehensive Immunophenotypic Analysis. Int. J. Surg. Pathol. 2020, 28, 440–446. [Google Scholar] [CrossRef]
- Thomas, M.; Bruant-Rodier, C.; Bodin, F.; Cribier, B.; Huther, M.; Dissaux, C. Why is it important to differentiate trichoblastic carcinomas (CT) from basal cell carcinomas (CBC). About 21 cases. Ann. Chir. Plast. Esthet. 2017, 62, 212–218. [Google Scholar] [CrossRef]
- Lepesant, P.; Crinquette, M.; Alkeraye, S.; Mirabel, X.; Dziwniel, V.; Cribier, B.; Mortier, L. Vismodegib induces significant clinical response in locally advanced trichoblastic carcinoma. Br. J. Derm. 2015, 173, 1059–1062. [Google Scholar] [CrossRef]
- Battistella, M.; Mateus, C.; Lassau, N.; Chami, L.; Boukoucha, M.; Duvillard, P.; Cribier, B.; Robert, C. Sunitinib efficacy in the treatment of metastatic skin adnexal carcinomas: Report of two patients with hidradenocarcinoma and trichoblastic carcinoma. J. Eur. Acad. Derm. Venereol. 2010, 24, 199–203. [Google Scholar] [CrossRef]
- Hamman, M.S.; Brian Jiang, S.I. Management of trichilemmal carcinoma: An update and comprehensive review of the literature. Derm. Surg. 2014, 40, 711–717. [Google Scholar] [CrossRef]
- Feng, Z.; Zhu, H.G.; Wang, L.Z.; Zheng, J.W.; Chen, W.T.; Zhang, Z.; Dong, W.; Qu, W.; Wang, Y.A. Tricholemmal carcinoma of the head and neck region: A report of 15 cases. Oncol. Lett. 2014, 7, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, K.; Sasaki, K.; Matsuda, M.; Hashimoto, M.; Eguchi, T.; Tomikawa, S.; Fujii, T.; Watanabe, G. A case of trichilemmal carcinoma with distant metastases in a kidney transplantation patient. Transpl. Proc. 2015, 47, 155–157. [Google Scholar] [CrossRef]
- Dalton, S.R.; LeBoit, P.E. Squamous cell carcinoma with clear cells: How often is there evidence of tricholemmal differentiation? Am. J. Derm. 2008, 30, 333–339. [Google Scholar] [CrossRef]
- Turnbull, N.; Ghumra, W.; Mudaliar, V.; Vella, J.; Sanders, D.S.A.; Taibjee, S.; Carr, R. CD34 and BerEP4 Are Helpful to Distinguish Basaloid Tricholemmoma From Basal Cell Carcinoma. Am. J. Derm. 2018, 40, 561–566. [Google Scholar] [CrossRef]
- Misago, N.; Toda, S.; Narisawa, Y. Tricholemmoma and clear cell squamous cell carcinoma (associated with Bowen’s disease): Immunohistochemical profile in comparison to normal hair follicles. Am. J. Derm. 2012, 34, 394–399. [Google Scholar] [CrossRef]
- Xu, B.; Wang, T.; Liao, Z. Surgical Treatment of Trichilemmal Carcinoma. World J. Oncol. 2018, 9, 141–144. [Google Scholar] [CrossRef]
- Jo, J.H.; Ko, H.C.; Jang, H.S.; Kim, M.B.; Oh, C.K.; Kwon, K.S. Infiltrative trichilemmal carcinoma treated with 5% imiquimod cream. Derm. Surg. 2005, 31, 973–976. [Google Scholar] [CrossRef]
- Parambeth, H.K.; Udhayam, N.; Agarwal, S.; Gupta, S.; Giridhar, P.; Rath, G.K. A large helmet-shaped proliferating trichilemmal tumor of the scalp: Is definitive radiotherapy the treatment? A case report. J. Egypt Natl. Cancer Inst. 2019, 31, 7. [Google Scholar] [CrossRef] [Green Version]
- Topkan, E. Adjuvant radiotherapy in management of trichilemmal carcinoma of left nasal alae with positive surgical margins: A case report. Case Rep. Clin. Pathol. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Roismann, M.; Freitas, R.R.; Ribeiro, L.C.; Montenegro, M.F.; Biasi, L.J.; Jung, J.E. Trichilemmal carcinoma: Case report. Bras. Derm. 2011, 86, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.S.; Sym, S.J.; Park, J.; Cho, E.K.; Ha, S.Y.; Shin, D.B.; Lee, J.H. Recurrent and metastatic trichilemmal carcinoma of the skin over the thigh: A case report. Cancer Res. Treat. 2010, 42, 176–179. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, S.M.; Zhang, G.H.; Chen, W.K.; Chen, S.W.; Wang, L.P.; Li, H.; Song, M. Survival study and clinicopathological evaluation of trichilemmal carcinoma. Mol. Clin. Oncol. 2013, 1, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Vanecek, T.; Nemcova, J.; Kacerovska, D.; Spagnolo, D.V.; Mukensnabl, P.; Michal, M. Spectrum of tumors with follicular differentiation in a patient with the clinical phenotype of multiple familial trichoepitheliomas: A clinicopathological and molecular biological study, including analysis of the CYLD and PTCH genes. Am. J. Derm. 2009, 31, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Pina, A.; Sauthier, P.; Rahimi, K. Vulvar trichoblastoma: Case report and literature review. J. Low Genit. Tract Dis. 2015, 19, e10–e12. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Nawrocki, S.; Hinther, K.; Khachemoune, A. Trichoblastomas Mimicking Basal Cell Carcinoma: The Importance of Identification and Differentiation. Cureus 2020, 12, e8272. [Google Scholar] [CrossRef]
- Cordoba, A.; Guerrero, D.; Larrinaga, B.; Iglesias, M.E.; Arrechea, M.A.; Yanguas, J.I. Bcl-2 and CD10 expression in the differential diagnosis of trichoblastoma, basal cell carcinoma, and basal cell carcinoma with follicular differentiation. Int. J. Derm. 2009, 48, 713–717. [Google Scholar] [CrossRef]
- Schulz, T.; Hartschuh, W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. An immunohistochemical study. J. Cutan. Pathol. 1997, 24, 14–24. [Google Scholar] [CrossRef]
- Izikson, L.; Bhan, A.; Zembowicz, A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am. J. Derm. 2005, 27, 91–95. [Google Scholar] [CrossRef]
- Jones, C.D.; Ho, W.; Robertson, B.F.; Gunn, E.; Morley, S. Pilomatrixoma: A Comprehensive Review of the Literature. Am. J. Derm. 2018, 40, 631–641. [Google Scholar] [CrossRef]
- Schwarz, Y.; Pitaro, J.; Waissbluth, S.; Daniel, S.J. Review of pediatric head and neck pilomatrixoma. Int. J. Pediatr. Otorhinolaryngol. 2016, 85, 148–153. [Google Scholar] [CrossRef]
- Swerlick, R.A.; Cooper, P.H.; Mackel, S.E. Rapid enlargement of pilomatricoma. J. Am. Acad. Dermatol. 1982, 7, 54–56. [Google Scholar] [CrossRef]
- Zhong, S.; Wang, L.; Mei, X.L. Desmoplastic trichilemmoma of the scalp: Case report and literature review of immunohistochemical staining features. J. Int. Med. Res. 2019, 47, 3918–3925. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.C.; Liau, J.Y.; Hong, J.L.; Cheng, Y.; Liao, Y.H.; Chen, J.S.; Sheen, Y.S.; Hong, J.B. Secondary neoplasms arising from nevus sebaceus: A retrospective study of 450 cases in Taiwan. J. Derm. 2016, 43, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zaballos, P.; Serrano, P.; Flores, G.; Bañuls, J.; Thomas, L.; Llambrich, A.; Castro, E.; Lallas, A.; Argenziano, G.; Zalaudek, I.; et al. Dermoscopy of tumours arising in naevus sebaceous: A morphological study of 58 cases. J. Eur. Acad. Derm. Venereol. 2015, 29, 2231–2237. [Google Scholar] [CrossRef]
- Pihlblad, M.; Chelnis, J.; Schaefer, D. Eyelid desmoplastic trichilemmoma: 2 case reports and review. Ophthalmic Plast. Reconstr. Surg. 2014, 30, e136–e138. [Google Scholar] [CrossRef]
- Stierman, S.; Chen, S.; Nuovo, G.; Thomas, J. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J. Cutan. Pathol. 2010, 37, 75–80. [Google Scholar] [CrossRef]
- Leonardi, C.L.; Zhu, W.Y.; Kinsey, W.H.; Penneys, N.S. Trichilemmomas are not associated with human papillomavirus DNA. J. Cutan. Pathol. 1991, 18, 193–197. [Google Scholar] [CrossRef]
- Maher, E.E.; Vidal, C.I. Trichilemmoma. Cutis 2015, 96, 81, 104–106. [Google Scholar]
- Hilliard, N.J.; Wakefield, D.N.; Krahl, D.; Sellheyer, K. p16 expression in conventional and desmoplastic trichilemmomas. Am. J. Derm. 2009, 31, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Tardío, J.C. CD34-reactive tumors of the skin. An updated review of an ever-growing list of lesions. J. Cutan. Pathol. 2009, 36, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, I.; Nishijima, S.; Kusumoto, K.; Senzaki, H.; Shikata, N.; Tsubura, A. Trichilemmoma: An immunohistochemical study of cytokeratins. Br. J. Derm. 2003, 149, 99–104. [Google Scholar] [CrossRef]
- Romero-Perez, D.; Garcia-Bustinduy, M.; Cribier, B. Clinicopathologic study of 90 cases of trichofolliculoma. J. Eur. Acad. Derm. Venereol. 2017, 31, e141–e142. [Google Scholar] [CrossRef]
- Al-Ghadeer, H.; Edward, D.P. Congenital Sebaceous Trichofolliculoma of the Upper Eyelid. Ophthalmic Plast Reconstr. Surg 2017, 33, S60–S61. [Google Scholar] [CrossRef]
- Ishii, N.; Kawaguchi, H.; Takahashi, K.; Nakajima, H. A case of congenital trichofolliculoma. J. Derm. 1992, 19, 195–196. [Google Scholar] [CrossRef]
- Misago, N.; Ansai, S.I.; Fukumoto, T.; Anan, T.; Kimura, T.; Nakao, T. Chronological changes in trichofolliculoma: Folliculosebaceous cystic hamartoma is not a very-late-stage trichofolliculoma. J. Derm. 2017, 44, 1050–1054. [Google Scholar] [CrossRef]
- Kapoor, A.G.; Vijitha, V.S.; Mittal, R. Trichofolliculoma of the Eyelid Margin: A Case Report and Review of Literature. Ophthalmic Plast. Reconstr. Surg. 2019, 35, e74–e76. [Google Scholar] [CrossRef]
- Bhawan, J. Pilar sheath acanthoma. A new benign follicular tumor. J. Cutan. Pathol. 1979, 6, 438–440. [Google Scholar] [CrossRef]
- Jo-Velasco, M.; Corrales-Rodriguez, A.; Frances-Rodriguez, L.; Alegria-Landa, V.; Erana-Tomas, I.; Rutten, A.; Requena, L. Plaque-Like Pilar Sheath Acanthoma: Histopathologic and Immunohistochemical Study of 3 Unusual Cases. Am. J. Derm. 2018, 40, 125–130. [Google Scholar] [CrossRef]
- Kushner, J.A.; Thomas, R.S.; Young, R.J. An unusual location of a pilar sheath acanthoma. Int. J. Trichology 2014, 6, 185–186. [Google Scholar] [CrossRef] [Green Version]
- Abbas, O.; Mahalingam, M. Tumor of the follicular infundibulum: An epidermal reaction pattern? Am. J. Derm. 2009, 31, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Weyers, W.; Horster, S.; Diaz-Cascajo, C. Tumor of follicular infundibulum is Basal cell carcinoma. Am. J. Derm. 2009, 31, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Baquerizo Nole, K.L.; Lopez-Garcia, D.R.; Teague, D.J.; Al Sayyah, A.; Mansoori, P.; Salim Al Alshehri, H.; Sangueza, O.P. Is Tumor of Follicular Infundibulum a Reaction to Dermal Scarring? Am. J. Derm. 2015, 37, 535–538. [Google Scholar] [CrossRef]
- Cheng, A.C.; Chang, Y.L.; Wu, Y.Y.; Hu, S.L.; Chuan, M.T. Multiple tumors of the follicular infundibulum. Derm. Surg. 2004, 30, 1246–1248. [Google Scholar] [CrossRef]
- Carlson, J.A.; Healy, K.; Slominski, A.; Mihm, M.C., Jr. Melanocytic matricoma: A report of two cases of a new entity. Am. J. Derm. 1999, 21, 344–349. [Google Scholar] [CrossRef]
- Zussman, J.; Sheth, S.; Ra, S.H.; Binder, S.W. Melanocytic matricoma with melanocytic atypia: Report of a unique case and review of the literature. Am. J. Derm. 2011, 33, 508–512. [Google Scholar] [CrossRef]
- Barrado-Solis, N.; Moles-Poveda, P.; Roca-Estelles, M.J.; Quecedo-Estebanez, E.; Gimeno-Carpio, E. Melanocytic matricoma with melanocytic atypia: Report of a new case. J. Eur. Acad. Derm. Venereol. 2016, 30, 859–860. [Google Scholar] [CrossRef]
- Ardakani, N.M.; Palmer, D.L.; Wood, B.A. Malignant Melanocytic Matricoma: A Report of 2 Cases and Review of the Literature. Am. J. Derm. 2016, 38, 33–38. [Google Scholar] [CrossRef]
- Michalova, K.; Kutzner, H.; Steiner, P.; Hadravsky, L.; Michal, M., Jr.; Michal, M.; Kazakov, D.V. Spindle Cell Predominant Trichodiscoma or Spindle Cell Lipoma With Adnexal Induction? A Study of 25 Cases, Revealing a Subset of Cases With RB1 Heterozygous Deletion in the Spindle Cell Stroma. Am. J. Derm. 2019, 41, 637–643. [Google Scholar] [CrossRef]
- Kutzner, H.; Requena, L.; Rutten, A.; Mentzel, T. Spindle cell predominant trichodiscoma: A fibrofolliculoma/trichodiscoma variant considered formerly to be a neurofollicular hamartoma: A clinicopathological and immunohistochemical analysis of 17 cases. Am. J. Derm. 2006, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zaballos, P.; Gomez-Martin, I.; Martin, J.M.; Banuls, J. Dermoscopy of Adnexal Tumors. Derm. Clin. 2018, 36, 397–412. [Google Scholar] [CrossRef]
- Mikhaeel, N.G.; Spittle, M.F. Malignant pilomatrixoma with multiple local recurrences and distant metastases: A case report and review of the literature. Clin. Oncol. 2001, 13, 386–389. [Google Scholar] [CrossRef]
- Hardisson, D.; Linares, M.D.; Cuevas-Santos, J.; Contreras, F. Pilomatrix carcinoma: A clinicopathologic study of six cases and review of the literature. Am. J. Derm. 2001, 23, 394–401. [Google Scholar] [CrossRef]
- Lobo, L.; Amonkar, A.D.; Dontamsetty, V.V. Malignant Proliferating Trichilemmal Tumour of the Scalp with Intra-Cranial Extension and Lung Metastasis-a Case Report. Indian J. Surg. 2016, 78, 493–495. [Google Scholar] [CrossRef] [Green Version]
- Okhremchuk, I.; Nguyen, A.T.; Fouet, B.; Morand, J.J. Trichoblastic Carcinosarcoma of the Skin: A Case Report and Literature Review. Am. J. Derm. 2018, 40, 917–919. [Google Scholar] [CrossRef]
- Laffay, L.; Depaepe, L.; d’Hombres, A.; Balme, B.; Thomas, L.; De Bari, B. Histological features and treatment approach of trichoblastic carcinomas: From a case report to a review of the literature. Tumori 2012, 98, 46e–49e. [Google Scholar] [CrossRef]
- Compaore, B.; Mosse, W.; Seka, E.; Kietga, G. Trichoblastic Carcinoma of the Upper Lip: A Case Report. Head Neck Cancer Res. 2020, 5, 9–12. [Google Scholar]
- Sajin, M.; Luchian, M.C.; Hodorogea Prisăcaru, A.; Dumitru, A.; Pătraşcu, O.M.; Costache, D.; Dumitrescu, D.; Oproiu, A.M.; Simionescu, O.; Costache, M. Trichilemmal carcinoma—A rare cutaneous malignancy: Report of two cases. Rom. J. Morphol. Embryol. 2014, 55, 687–691. [Google Scholar]





| Cancer Type | Genetic Abnormalities | Reference | ||
|---|---|---|---|---|
| Mutation | Expression Downregulated | Expression Upregulated | ||
| Pilomatrical carcinoma | CTNNB1, CDX2, LEF-1 | CDX2, LEF-1 | [9,10] | |
| Proliferating trichilemmal tumor | TP53, PI3KCA | TP53 | PIK3 | [11,12,13] |
| Trichoblastic carcinoma/carcinosarcoma | TP53, CDKN2A | |||
| Trichilemmal carcinoma | TP53, NF1, TOP1, PTEN | PTEN, TP53 | TOP1 | [14] |
| Trichoblastoma | CYLD, CTNNB1, HRAS, PTCH1 | HRAS | [10,15,16] | |
| Pilomatrixoma | CTNNB1, BCL2, APC, CTNNB1 | [6,17,18,19,20,21] | ||
| Trichilemmoma | HRAS, PTEN | HRAS | [22] | |
| Trichofolliculoma | BMP, PYGO2 (only experimental models) | [23] | ||
| Pilar sheath acanthoma | ND | |||
| Tumor of the follicular infundibulum | ND | |||
| Melanocytic matricoma | ND | |||
| Spindle cell-predominant trichodiscoma | ND | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Płachta, I.; Kleibert, M.; Czarnecka, A.M.; Spałek, M.; Szumera-Ciećkiewicz, A.; Rutkowski, P. Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation. Int. J. Mol. Sci. 2021, 22, 4759. https://doi.org/10.3390/ijms22094759
Płachta I, Kleibert M, Czarnecka AM, Spałek M, Szumera-Ciećkiewicz A, Rutkowski P. Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation. International Journal of Molecular Sciences. 2021; 22(9):4759. https://doi.org/10.3390/ijms22094759
Chicago/Turabian StylePłachta, Iga, Marcin Kleibert, Anna M. Czarnecka, Mateusz Spałek, Anna Szumera-Ciećkiewicz, and Piotr Rutkowski. 2021. "Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation" International Journal of Molecular Sciences 22, no. 9: 4759. https://doi.org/10.3390/ijms22094759
APA StylePłachta, I., Kleibert, M., Czarnecka, A. M., Spałek, M., Szumera-Ciećkiewicz, A., & Rutkowski, P. (2021). Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Follicular Differentiation. International Journal of Molecular Sciences, 22(9), 4759. https://doi.org/10.3390/ijms22094759