
The Impact of microRNAs in Renin–Angiotensin-System-Induced Cardiac Remodelling


Abstract
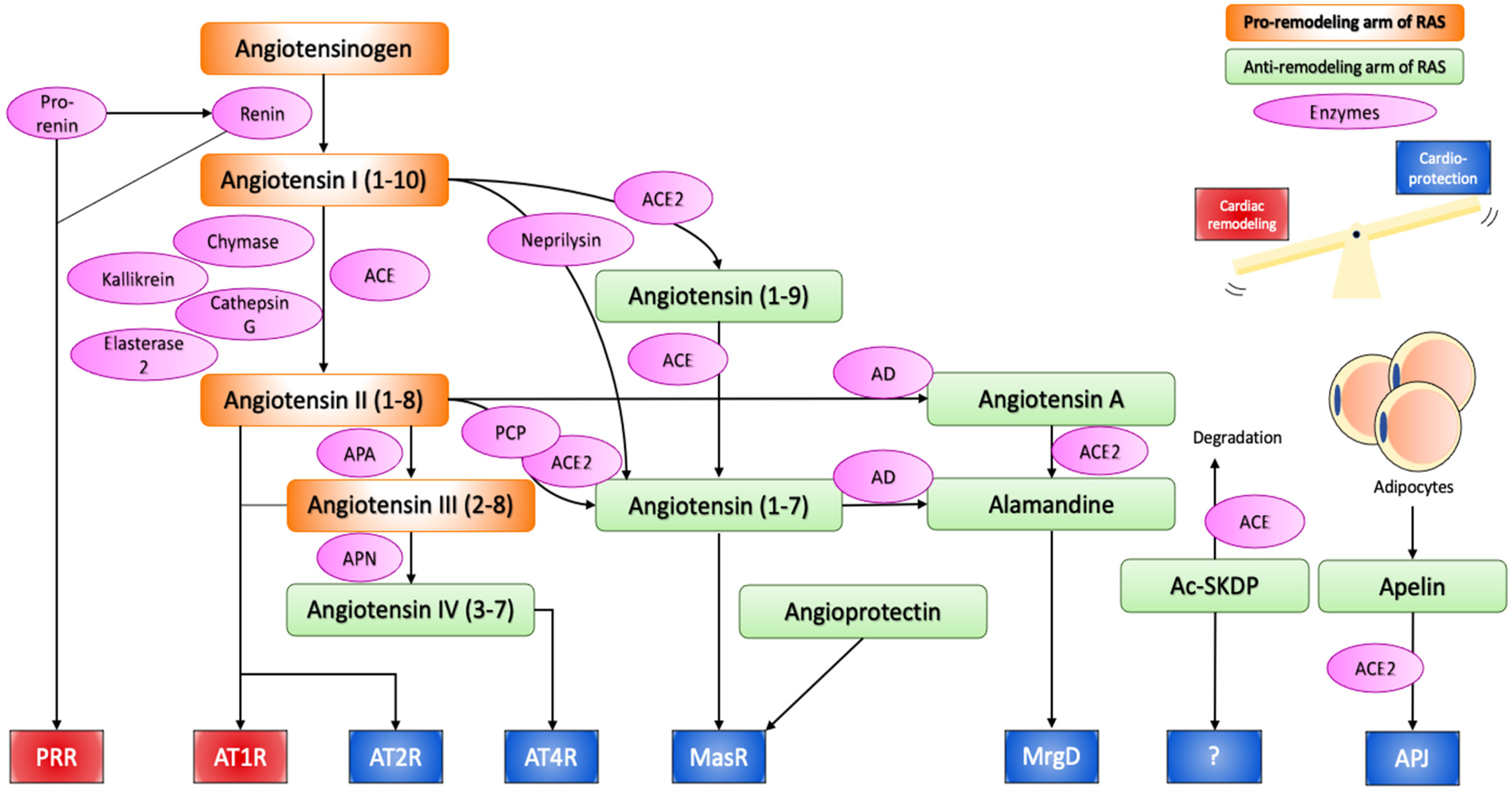
:1. The Renin–Angiotensin System
1.1. Classical Pathways
1.2. Alternative Pathways of RAS
1.3. Local RAS
1.4. Intracellular or Intracrine RAS
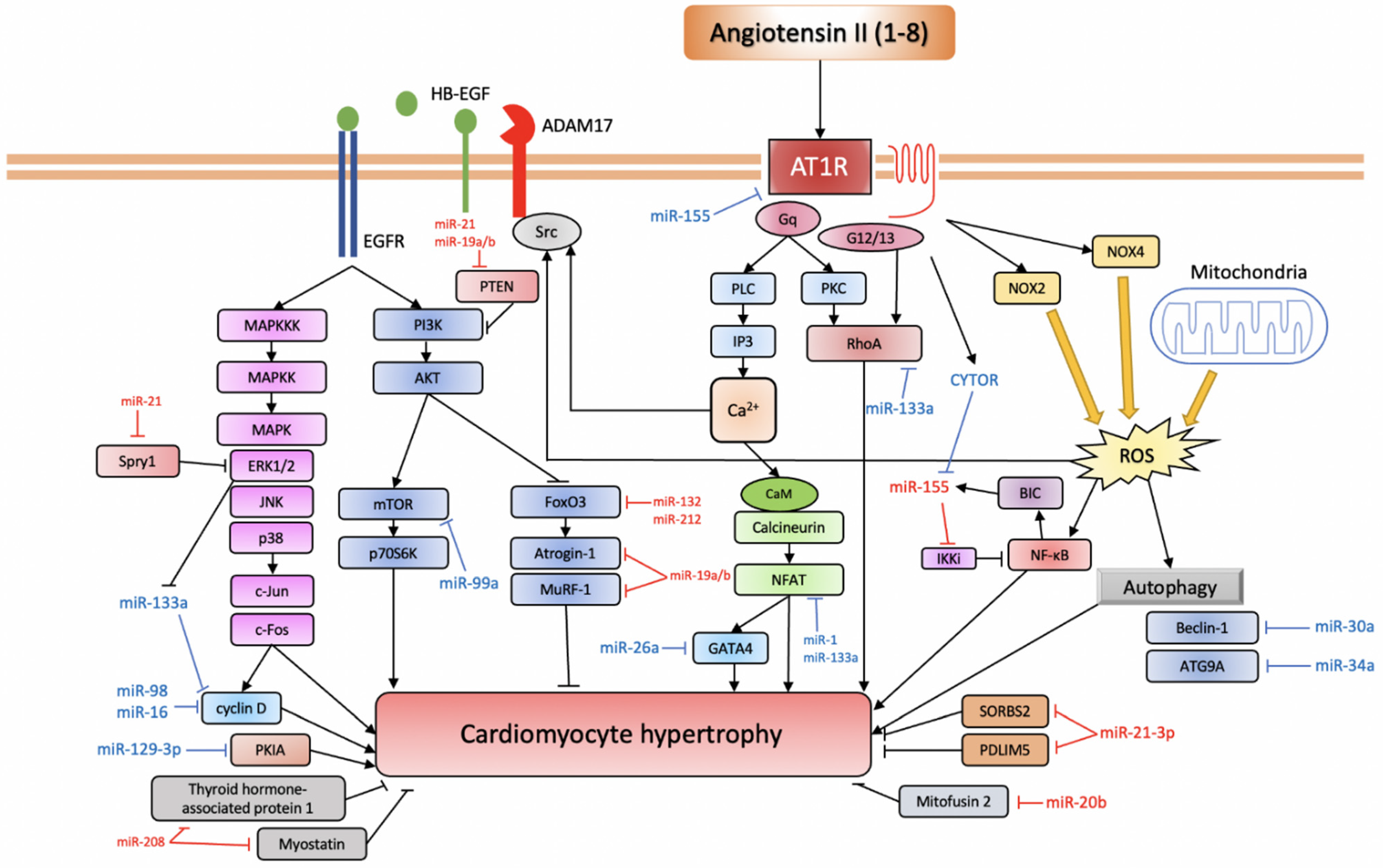
2. Angiotensin II Signalling and Cardiac Remodelling
3. microRNAs in the Regulation of RAS-Induced Cardiac Remodelling
3.1. miRNAs Involved in Ang II-Induced Cardiac Hypertrophy
3.1.1. Pro-Hypertrophic miRNAs
miR-155
miR-208
miR-132/212
miR-21
miR-410, miR-495
miR-19a/b
miR-20b
3.1.2. Anti-Hypertrophic miRNAs
miR-21-3p, miR-26a
miR-16, miR-98
miR-30a, miR-34a
miR-133a, miR-1
miR-99a, miR-101
miR-129-3p
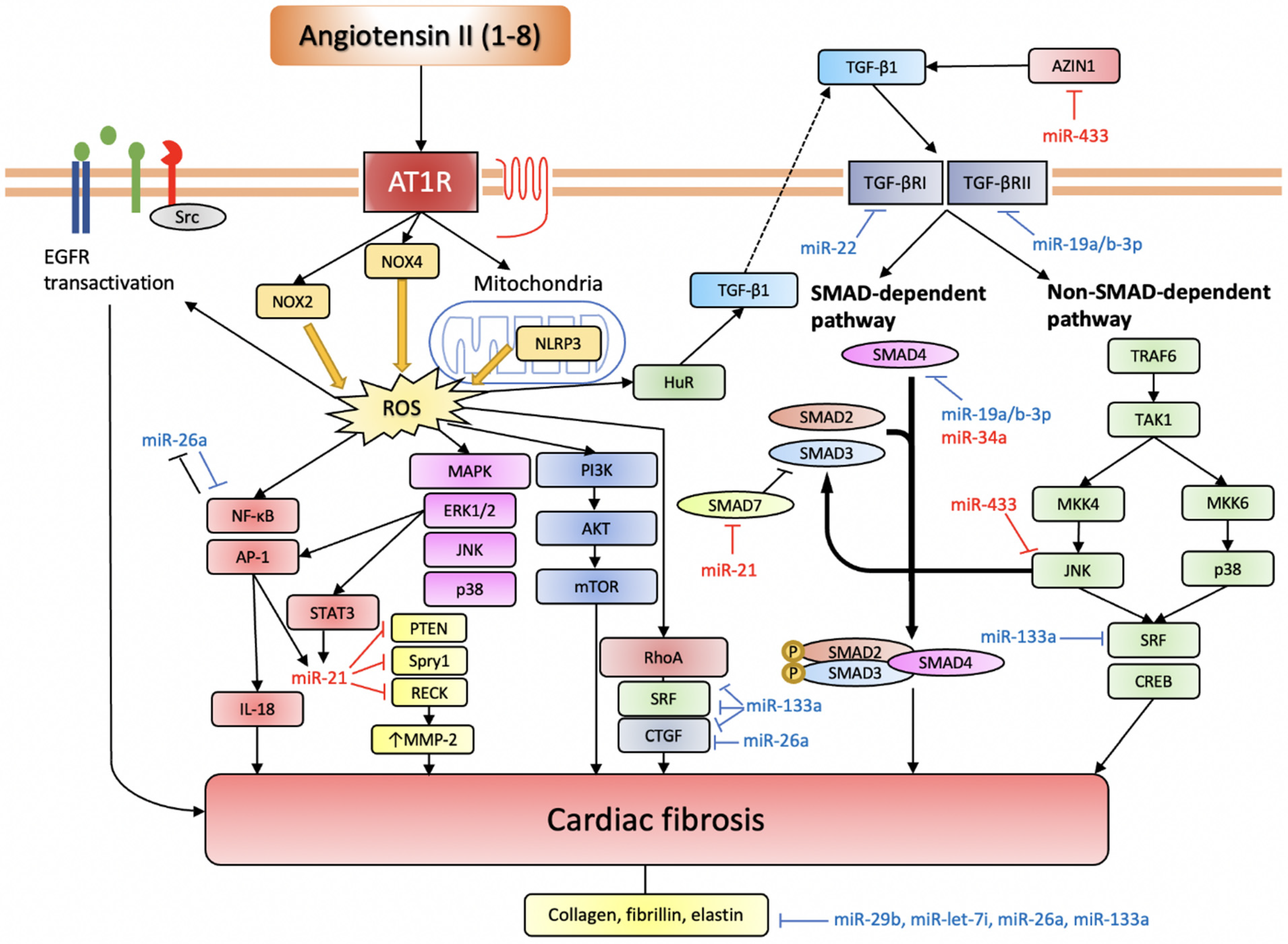
3.2. miRNAs in Ang II-Induced Cardiac Fibrosis
3.2.1. Pro-Fibrotic miRNAs
miR-21
miR-433
miR-503
miR-34a
miR-155
3.2.2. Anti-Fibrotic miRNAs
miR-26a
miR-133a
miR-19a/b-3p
miR-29b
miR-22
miR-let-7i
3.3. miRNAs That Directly Target RAS Components
3.3.1. miR-181a
3.3.2. miR-143/145 Cluster
3.3.3. miR-483-3p
3.3.4. miR-766
3.3.5. miR-125b

{kind=link}
{kind=link}
{kind=link}
| miRNA | Target | Function | Source |
|---|---|---|---|
| miR-155 | Socs1, Jarid2, AT1R, IKKi eNOS | Promote pro-hypertrophic and pro-fibrotic inflammation, but anti-hypertrophic through targeting AT1R | [90,91,92,95,97] |
| miR-208 | Thyroid hormone-associated protein 1, myostatin, NLK | Promote cardiac hypertrophy and arrhythmia, involved in apoptosis | [104,109] |
| miR-132/212 | FoxO3, AT1R, AC, JAK2, PKC, cJUN, SOD2, EGR1 | Promote cardiac hypertrophy, inhibit autophagy and apoptosis | [112,114] |
| miR-21 | Spry1, PTEN, SMAD7, TGFBR2, RECK | Promote cardiac hypertrophy and fibrosis | [116,201,202,203,204,206,210,212] |
| miR-410/495 | MDM2, MET, MTA3, MTA1A, SOX9, BMI1,TBC1D9, PPX3, MEIS1, ATP7, PTP4A3, SMR3B | Promote cardiomyocyte hypertrophy and proliferation | [118] |
| miR-19a/b | Atrogin-1, MuRF-1, PTEN | Promote cardiac hypertrophy, inhibit apoptosis, reduce CTGF expression | [119,120,121] |
| miR-20b, 20b-5p | Mitofusin2, SMAD7 | Promote cardiac hypertrophy and ventricular remodelling | [123,125] |
| miR-21-3p | SORBS2, PDLIM5, HDAC8 | Involved in cardiac hypertrophy (evidence for both pro- and anti-hypertrophic effects) | [126,128] |
| miR-26a | GATA4, CTGF, collagen I | Inhibit cardiac hypertrophy and cardiac fibrosis | [127,228] |
| miR-16 | Cyclin D1, D2, E1 | Inhibit cardiac hypertrophy | [129] |
| miR-98 | Cyclin D2 | Inhibits cardiac hypertrophy | [130] |
| miR-30a | Beclin-1 | Inhibits cardiac autophagy and hypertrophy | [131] |
| miR-34a | ATG9A, SMAD4 | Inhibit cardiac autophagy and hypertrophy, promote cardiac fibrosis | [132,224] |
| miR-133a | Angiotensinogen, SRF, cyclin D2, caspase 9, CTGF, RhoA, Cdc42, Nel-A/WHSC2, collagen 1α1, NFATc4 | Inhibit cardiac hypertrophy and cardiac fibrosis | [135,141,142,143,144,145,146,147] |
| miR-1 | HDAC4, NFATc3, MCU, CDK6, IGF-1, IGF-1R | Inhibit cardiac hypertrophy | [141,152,153] |
| miR-99a | mTOR1/2 | Inhibits cardiac hypertrophy | [157] |
| miR-101 | Rab1a | Inhibits cardiac hypertrophy | [158] |
| miR-129-3p | PKIA | Inhibits cardiac hypertrophy | [159] |
| miR-433 | JNK1, AZIN1 | Promote cardiac fibrosis | [215] |
| miR-503 | Apelin-13 | Promotes cardiac fibrosis | [216] |
| miR-19a/b-3p | TGFBR2, SMAD4 | Inhibit cardiac fibrosis | [230,231,232] |
| miR-29b | Collagen, fibrillin, elastin | Inhibit cardiac fibrosis | [233,234,235] |
| miR-22 | TGFBR1 | Inhibits cardiac fibrosis | [237] |
| miR-let-7i | IL-6, collagens | Inhibit cardiac inflammation and fibrosis | [238] |
| miR-181a | REN, AIFM1 | Reduce blood pressure | [239] |
| miR-143/145 | KLF4/5, myocardin, ELK-1 | Cardiogenesis, maintenance of VSMC phenotype | [243,245,246] |
| miR-145, 27a/b | ACE | RAS modulation | [246,247] |
| miR-143, 421 | ACE2 | RAS modulation | [34,250,251] |
| miR-483-3p | ACE, ACE2, AT2R | RAS modulation | [252] |
| miR-766 | Cyp11B2 | Reduces aldosterone synthase | [253] |
| miR-125b | ACE2 | RAS modulation | [254] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goodman, L.S.; Brunton, L.L.; Chabner, B.; Knollmann, B.C. Goodman & Gilman’s Pharmacological Basis of Therapeutics; McGraw-Hill: New York, NY, USA, 2011; pp. 721–741. [Google Scholar]
- Von Lueder, T.G.; Krum, H. RAAS inhibitors and cardiovascular protection in large scale trials. Cardiovasc. Drugs Ther. 2013, 27, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova-Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediators Inflamm. 2014, 2014, 703175:1–703175:10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simko, F.; Pechanova, O.; Krajcirovicova, K.; Matuskova, J.; Pelouch, V.; Adamcova, M.; Paulis, L. Effects of captopril, spironolactone, and simvastatin on the cardiovascular system of non-diseased Wistar rats. Int. J. Cardiol. 2015, 190, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Celec, P.; Tothova, L.; Vrankova, S.; Balazova, L.; Zorad, S.; et al. Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril. Int. J. Mol. Sci. 2017, 18, 1612. [Google Scholar] [CrossRef] [Green Version]
- Simko, F.; Baka, T.; Poglitsch, M.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Zorad, S.; Adamcova, M.; Paulis, L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. Int. J. Mol. Sci. 2018, 19, 3017. [Google Scholar] [CrossRef] [Green Version]
- Simko, F.; Hrenak, J.; Adamcova, M.; Paulis, L. Renin-Angiotensin-Aldosterone System: Friend or Foe—The Matter of Balance. Insight on History, Therapeutic Implications and COVID-19 Interactions. Int. J. Mol. Sci. 2021, 22, 3217. [Google Scholar] [CrossRef]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell. Signal. 2016, 1, 111. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lorenzo, O.; Rupérez, M.; König, S.; Wittig, B.; Egido, J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: Molecular mechanisms. Circ. Res. 2000, 86, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieffer, B.; Wirger, A.; Meybrunn, M.; Seitz, S.; Holtz, J.; Riede, U.N.; Drexler, H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994, 89, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J. Hypertens. Suppl. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; Lother, H.; Abdel-tawab, A.M.; Quitterer, U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 2001, 276, 39721–39726. [Google Scholar] [CrossRef] [Green Version]
- Uehara, Y.; Miura, S.; Yahiro, E.; Saku, K. Non-ACE pathway-induced angiotensin II production. Curr. Pharm. Des. 2013, 19, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J.; Fabryová, M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: Pathophysiological consideration of the unresolved battle. Cardiovasc. Drugs Ther. 2003, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Hrenak, J.; Simko, F. Renin-Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, E8038. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Chappell, M.C.; Tallant, E.A.; Brosnihan, K.B.; Diz, D.I. Counterregulatory actions of angiotensin-(1-7). Hypertension 1997, 30, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Grobe, J.L.; Vazquez, J.; Stewart, J.M.; Mecca, A.P.; Katovich, M.J.; Ferrario, C.M.; Raizada, M.K. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp. Physiol. 2005, 90, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Santos, R.A.; Ferreira, A.J.; Simões E Silva, A.C. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp. Physiol. 2008, 93, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M. Angiotensin-converting enzyme 2 and angiotensin-(1-7): An evolving story in cardiovascular regulation. Hypertension 2006, 47, 515–521. [Google Scholar] [CrossRef]
- Schindler, C.; Bramlage, P.; Kirch, W.; Ferrario, C.M. Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc. Health Risk Manag. 2007, 3, 125–137. [Google Scholar] [PubMed]
- Walters, P.E.; Gaspari, T.A.; Widdop, R.E. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 2005, 45, 960–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-Protein-Coupled Receptor MrgD Is a Receptor for Angiotensin-(1-7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase A. Hypertension 2016, 68, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Schleifenbaum, J. Alamandine and Its Receptor MrgD Pair Up to Join the Protective Arm of the Renin-Angiotensin System. Front. Med. (Lausanne) 2019, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, V.; Vanholder, R.; van der Giet, M.; Tölle, M.; Karadogan, S.; Gobom, J.; Furkert, J.; Oksche, A.; Krause, E.; Tran, T.N.; et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, V.; Tölle, M.; Santos, R.A.; Günthner, T.; Krause, E.; Beyermann, M.; Welker, P.; Bader, M.; Pinheiro, S.V.; Sampaio, W.O.; et al. Angioprotectin: An angiotensin II-like peptide causing vasodilatory effects. FASEB J. 2011, 25, 2987–2995. [Google Scholar] [CrossRef]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.; Simpson, R.J.; Connolly, L.M.; et al. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.W.; Miller-Wing, A.V.; Shaffer, M.J.; Higginson, C.; Wright, D.E.; Hanesworth, J.M.; Harding, J.W. Angiotensin II(3-8) (ANG IV) hippocampal binding: Potential role in the facilitation of memory. Brain Res. Bull. 1993, 32, 497–502. [Google Scholar] [CrossRef]
- Hrenak, J.; Paulis, L.; Simko, F. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP): Potential target molecule in research of heart, kidney and brain. Curr. Pharm. Des. 2015, 21, 5135–5143. [Google Scholar] [CrossRef]
- Chen, L.J.; Xu, R.; Yu, H.M.; Chang, Q.; Zhong, J.C. The ACE2/Apelin Signaling, MicroRNAs, and Hypertension. Int. J. Hypertens. 2015, 2015, 896861. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Tang, Z.B.; Liu, L.J.; Qian, H.; Tang, S.L.; Zhang, D.W.; Tian, G.P.; Tang, C.K. Apelin and its receptor APJ in cardiovascular diseases. Clin. Chim. Acta 2014, 428, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8. [Google Scholar]
- Nguyen, G.; Danser, A.H. Prorenin and (pro)renin receptor: A review of available data from in vitro studies and experimental models in rodents. Exp. Physiol. 2008, 93, 557–563. [Google Scholar] [CrossRef]
- Mahmud, H.; Silljé, H.H.; Cannon, M.V.; van Gilst, W.H.; de Boer, R.A. Regulation of the (pro)renin-renin receptor in cardiac remodelling. J. Cell Mol. Med. 2012, 16, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, A.; Yatabe, M.S. The (pro)renin receptor in health and disease. Nat. Rev. Nephrol. 2019, 15, 693–712. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, E14. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Le, B.; Bhat, V.B.; Baker, K.M.; Kumar, R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H939–H948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Singh, V.P.; Baker, K.M. The intracellular renin-angiotensin system: A new paradigm. Trends Endocrinol. Metab. 2007, 18, 208–214. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, V.P.; Baker, K.M. The intracellular renin-angiotensin system in the heart. Curr. Hypertens. Rep. 2009, 11, 104–110. [Google Scholar] [CrossRef]
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1675–H1684. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Crabos, M.; Roth, M.; Hahn, A.W.; Erne, P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J. Clin. Investig. 1994, 93, 2372–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkubo, N.; Matsubara, H.; Nozawa, Y.; Mori, Y.; Murasawa, S.; Kijima, K.; Maruyama, K.; Masaki, H.; Tsutumi, Y.; Shibazaki, Y.; et al. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation 1997, 96, 3954–3962. [Google Scholar] [CrossRef]
- Senbonmatsu, T.; Ichihara, S.; Price, E.; Gaffney, F.A.; Inagami, T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J. Clin. Investig. 2000, 106, R25–R29. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leri, A.; Claudio, P.P.; Li, Q.; Wang, X.; Reiss, K.; Wang, S.; Malhotra, A.; Kajstura, J.; Anversa, P. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J. Clin. Investig. 1998, 101, 1326–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef]
- Lunde, I.G.; Kvaløy, H.; Austbø, B.; Christensen, G.; Carlson, C.R. Angiotensin II and norepinephrine activate specific calcineurin-dependent NFAT transcription factor isoforms in cardiomyocytes. J. Appl. Physiol. (1985) 2011, 111, 1278–1289. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E. The art of microRNA research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. Biomed. Res. Int. 2017, 2017, 1278436. [Google Scholar] [CrossRef] [Green Version]
- Deiuliis, J.; Mihai, G.; Zhang, J.; Taslim, C.; Varghese, J.J.; Maiseyeu, A.; Huang, K.; Rajagopalan, S. Renin-sensitive microRNAs correlate with atherosclerosis plaque progression. J. Hum. Hypertens. 2014, 28, 251–258. [Google Scholar] [CrossRef]
- Butterworth, M.B. Role of microRNAs in aldosterone signaling. Curr. Opin. Nephrol. Hypertens. 2018, 27, 390–394. [Google Scholar] [CrossRef]
- Butterworth, M.B. Non-coding RNAs and the mineralocorticoid receptor in the kidney. Mol. Cell Endocrinol. 2021, 521, 111115. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Takefuji, M.; Wirth, A.; Lukasova, M.; Takefuji, S.; Boettger, T.; Braun, T.; Althoff, T.; Offermanns, S.; Wettschureck, N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation 2012, 126, 1972–1982. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 627–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Li, Y.; Xiao, C.; Du, J. Angiotensin II induces inflammation leading to cardiac remodeling. Front. Biosci. (Landmark Ed.) 2012, 17, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Ichiki, T.; Ito, K.; Takeshita, A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 1999, 34, 118–125. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Yu, H.; Zhang, Y.; Dai, Y.; Ning, C.; Tao, L.; Sun, H.; Kellems, R.E.; Blackburn, M.R.; et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 2012, 59, 136–144. [Google Scholar] [CrossRef]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Sriramula, S.; Francis, J. Tumor Necrosis Factor-Alpha Is Essential for Angiotensin II-Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS ONE 2015, 10, e0138372. [Google Scholar] [CrossRef]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Liu, X.; Xu, J.; Weng, L.; Ren, J.; Ge, J.; Zou, Y. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J. Cell. Mol. Med. 2016, 20, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Kishore, R.; Krishnamurthy, P.; Garikipati, V.N.; Benedict, C.; Nickoloff, E.; Khan, M.; Johnson, J.; Gumpert, A.M.; Koch, W.J.; Verma, S.K. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J. Mol. Cell. Cardiol. 2015, 89, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Wang, X.; McBride, J.; Fewell, C.; Flemington, E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2008, 283, 2654–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elton, T.S.; Selemon, H.; Elton, S.M.; Parinandi, N.L. Regulation of the MIR155 host gene in physiological and pathological processes. Gene 2013, 532, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.; Papavasiliou, F.N. Shhh! Silencing by microRNA-155. Philos. Trans. R Soc. Lond. B Biol. Sci. 2009, 364, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; van Leeuwen, R.E.; Custers, K.; Peters, T.; Hazebroek, M.; Stöger, L.; Wijnands, E.; et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, H.Y.; Chen, J.; Kataoka, M.; Huang, Z.P.; Ding, J.; Yan, J.; Hu, X.; Wang, D.Z. Loss of MicroRNA-155 protects the heart from pathological cardiac hypertrophy. Circ. Res. 2014, 114, 1585–1595. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, J.; Chen, Q.; Wu, Q.; Deng, W.; Zhou, H.; Shen, D. Long non-coding RNA cytoskeleton regulator RNA (CYTOR) modulates pathological cardiac hypertrophy through miR-155-mediated IKKi signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Qin, L.; Peng, Y.; Bai, W.; Wang, Z. Exosomes Derived from Hypertrophic Cardiomyocytes Induce Inflammation in Macrophages via miR-155 Mediated MAPK Pathway. Front. Immunol. 2020, 11, 606045. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y.; Cao, Z.; Tong, X.Z.; Xie, H.Q.; Luo, T.; Hua, X.P.; Wang, H.Q. miR-155 functions downstream of angiotensin II receptor subtype 1 and calcineurin to regulate cardiac hypertrophy. Exp. Ther. Med. 2016, 12, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Chen, S.; Liu, X.; Lin, L.; Huang, X.; Guo, Z.; Liu, J.; Wang, Y.; Yuan, W.; et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 2011, 215, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, T.; Jiang, F.; Liu, C.; Zhao, X.; Gao, Y.; Wang, H.; Liu, Z. microRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int. J. Mol. Med. 2011, 27, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.X.; Zeng, D.Y.; Li, R.T.; Pang, R.P.; Yang, H.; Hu, Y.L.; Zhang, Q.; Jiang, Y.; Huang, L.Y.; Tang, Y.B.; et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Alexy, T.; Rooney, K.; Weber, M.; Gray, W.D.; Searles, C.D. TNF-α alters the release and transfer of microparticle-encapsulated miRNAs from endothelial cells. Physiol. Genom. 2014, 46, 833–840. [Google Scholar] [CrossRef]
- Zheng, L.; Xu, C.C.; Chen, W.D.; Shen, W.L.; Ruan, C.C.; Zhu, L.M.; Zhu, D.L.; Gao, P.J. MicroRNA-155 regulates angiotensin II type 1 receptor expression and phenotypic differentiation in vascular adventitial fibroblasts. Biochem. Biophys. Res. Commun. 2010, 400, 483–488. [Google Scholar] [CrossRef]
- Yang, L.X.; Liu, G.; Zhu, G.F.; Liu, H.; Guo, R.W.; Qi, F.; Zou, J.H. MicroRNA-155 inhibits angiotensin II-induced vascular smooth muscle cell proliferation. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Meng, H.; Jiang, C.; Yang, S.; Cui, F.; Yang, P. Differential microRNA Expression and Regulation in the Rat Model of Post-Infarction Heart Failure. PLoS ONE 2016, 11, e0160920. [Google Scholar] [CrossRef]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimoto, Y.; Tanaka, M.; Kamiguchi, H.; Hayashi, H.; Ochiai, E.; Osawa, M. MicroRNA deep sequencing reveals chamber-specific miR-208 family expression patterns in the human heart. Int. J. Cardiol. 2016, 211, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.D.; Meng, A.C.; Zhu, Q.; Jia, R.Y.; Kong, Q.Z. Effect of microRNA-208a on mitochondrial apoptosis of cardiomyocytes of neonatal rats. Asian Pac. J. Trop. Med. 2015, 8, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Tony, H.; Yu, K.; Qiutang, Z. MicroRNA-208a Silencing Attenuates Doxorubicin Induced Myocyte Apoptosis and Cardiac Dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 597032. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, C.; Liu, X.; Yang, P. Ghrelin Alleviates Angiotensin II-Induced H9c2 Apoptosis: Impact of the miR-208 Family. Med. Sci. Monit. 2018, 24, 6707–6716. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, Y.; He, Y.; Huang, C.; Meng, X.; Li, J. MicroRNA-208a Potentiates Angiotensin II-triggered Cardiac Myoblasts Apoptosis via Inhibiting Nemo-like Kinase (NLK). Curr. Pharm. Des. 2016, 22, 4868–4875. [Google Scholar] [CrossRef]
- Rana, I.; Velkoska, E.; Patel, S.K.; Burrell, L.M.; Charchar, F.J. MicroRNAs mediate the cardioprotective effect of angiotensin-converting enzyme inhibition in acute kidney injury. Am. J. Physiol. Renal Physiol. 2015, 309, F943–F954. [Google Scholar] [CrossRef] [Green Version]
- Jeppesen, P.L.; Christensen, G.L.; Schneider, M.; Nossent, A.Y.; Jensen, H.B.; Andersen, D.C.; Eskildsen, T.; Gammeltoft, S.; Hansen, J.L.; Sheikh, S.P. Angiotensin II type 1 receptor signalling regulates microRNA differentially in cardiac fibroblasts and myocytes. Br. J. Pharmacol. 2011, 164, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, T.V.; Jeppesen, P.L.; Schneider, M.; Nossent, A.Y.; Sandberg, M.B.; Hansen, P.B.; Jensen, C.H.; Hansen, M.L.; Marcussen, N.; Rasmussen, L.M.; et al. Angiotensin II regulates microRNA-132/-212 in hypertensive rats and humans. Int. J. Mol. Sci. 2013, 14, 11190–11207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskildsen, T.V.; Schneider, M.; Sandberg, M.B.; Skov, V.; Brønnum, H.; Thomassen, M.; Kruse, T.A.; Andersen, D.C.; Sheikh, S.P. The microRNA-132/212 family fine-tunes multiple targets in Angiotensin II signalling in cardiac fibroblasts. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 1288–1297. [Google Scholar] [CrossRef]
- Foinquinos, A.; Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Gyöngyösi, M.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020, 11, 633. [Google Scholar] [CrossRef]
- Cheng, Y.; Ji, R.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a role in cardiac hypertrophy. Am. J. Pathol. 2007, 170, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duygu, B.; Da Costa Martins, P.A. miR-21: A star player in cardiac hypertrophy. Cardiovasc. Res. 2015, 105, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.L.; Maruyama, S.; Sano, S.; Accorsi, A.; Girgenrath, M.; Walsh, K.; Naya, F.J. miR-410 and miR-495 Are Dynamically Regulated in Diverse Cardiomyopathies and Their Inhibition Attenuates Pathological Hypertrophy. PLoS ONE 2016, 11, e0151515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.W.; Ryu, J.Y.; Kim, J.O.; Kwon, E.J.; Kim, D.H. The miR-19a/b family positively regulates cardiomyocyte hypertrophy by targeting atrogin-1 and MuRF-1. Biochem. J. 2014, 457, 151–162. [Google Scholar] [CrossRef]
- Gao, F.; Kataoka, M.; Liu, N.; Liang, T.; Huang, Z.P.; Gu, F.; Ding, J.; Liu, J.; Zhang, F.; Ma, Q.; et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun. 2019, 10, 1802. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Liu, T.W.; Wang, Z.; Jiao, Z.Y.; Cai, J.; Chi, H.J.; Yang, X.C. Downregulation of microRNA-19b contributes to angiotensin II-induced overexpression of connective tissue growth factor in cardiomyocytes. Cardiology 2014, 127, 114–120. [Google Scholar] [CrossRef]
- Liu, K.; Hao, Q.; Wei, J.; Li, G.H.; Wu, Y.; Zhao, Y.F. MicroRNA-19a/b-3p protect the heart from hypertension-induced pathological cardiac hypertrophy through PDE5A. J. Hypertens. 2018, 36, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cheng, R.; Liang, C.; Yao, Y.; Zhang, W.; Zhang, J.; Zhang, M.; Li, B.; Xu, C.; Zhang, R. MicroRNA-20b Promotes Cardiac Hypertrophy by the Inhibition of Mitofusin 2-Mediated Inter-organelle Ca2+ Cross-Talk. Mol. Ther. Nucleic Acids 2020, 19, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.H.; Huang, X.R.; Zhang, Y.; Li, Y.Q.; Chen, H.Y.; Yan, B.P.; Yu, C.M.; Lan, H.Y. Smad7 inhibits angiotensin II-induced hypertensive cardiac remodelling. Cardiovasc. Res. 2013, 99, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.G.; Yao, H.; Xie, R.S.; Gong, C.L.; Tian, Y. MicroRNA-20b-5p promotes ventricular remodeling by targeting the TGF-β/Smad signaling pathway in a rat model of ischemia-reperfusion injury. Int. J. Mol. Med. 2018, 42, 975–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Chen, C.; Gong, W.; Yin, Z.; Zhou, L.; Chaugai, S.; Wang, D.W. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc. Res. 2015, 105, 340–352. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Xiao, W. MicroRNA-26a protects against cardiac hypertrophy via inhibiting GATA4 in rat model and cultured cardiomyocytes. Mol. Med. Rep. 2016, 14, 2860–2866. [Google Scholar] [CrossRef] [Green Version]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Huang, S.; Zou, X.; Zhu, J.N.; Fu, Y.H.; Lin, Q.X.; Liang, Y.Y.; Deng, C.Y.; Kuang, S.J.; Zhang, M.Z.; Liao, Y.L.; et al. Attenuation of microRNA-16 derepresses the cyclins D1, D2 and E1 to provoke cardiomyocyte hypertrophy. J. Cell. Mol. Med. 2015, 19, 608–619. [Google Scholar] [CrossRef]
- Yang, Y.; Ago, T.; Zhai, P.; Abdellatif, M.; Sadoshima, J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ. Res. 2011, 108, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Zhong, Y.; Cheng, C.; Liu, B.; Wang, L.; Li, A.; Xiong, L.; Liu, S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS ONE 2013, 8, e53950. [Google Scholar] [CrossRef]
- Huang, J.; Sun, W.; Huang, H.; Ye, J.; Pan, W.; Zhong, Y.; Cheng, C.; You, X.; Liu, B.; Xiong, L.; et al. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS ONE 2014, 9, e94382. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhao, J.; Tuazon, J.P.; Borlongan, C.V.; Yu, G. MicroRNA-133a and Myocardial Infarction. Cell Transplant. 2019, 28, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Leptidis, S.; El Azzouzi, H.; Lok, S.I.; de Weger, R.; Olieslagers, S.; Olieslagers, S.; Kisters, N.; Silva, G.J.; Heymans, S.; Cuppen, E.; et al. A deep sequencing approach to uncover the miRNOME in the human heart. PLoS ONE 2013, 8, e57800. [Google Scholar] [CrossRef] [Green Version]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Puthanveetil, P.; Feng, B.; Matkovich, S.J.; Dorn, G.W.; Chakrabarti, S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 2014, 18, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Chen, S.; George, B.; Feng, Q.; Chakrabarti, S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab. Res. Rev. 2010, 26, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Matkovich, S.J.; Wang, W.; Tu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Condorelli, G.; Diwan, A.; Nerbonne, J.M.; Dorn, G.W. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ. Res. 2010, 106, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, D.; Hong, C.; Chen, I.Y.; Lypowy, J.; Abdellatif, M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef] [Green Version]
- Abdellatif, M. The role of microRNA-133 in cardiac hypertrophy uncovered. Circ. Res. 2010, 106, 16–18. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef] [Green Version]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoldi, G.; Di Gioia, C.R.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. MiR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef]
- Li, Q.; Lin, X.; Yang, X.; Chang, J. NFATc4 is negatively regulated in miR-133a-mediated cardiomyocyte hypertrophic repression. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1340–H1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.M.; Nandi, S.S.; Zheng, H.; Mishra, P.K.; Patel, K.P. A novel role for miR-133a in centrally mediated activation of the renin-angiotensin system in congestive heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H968–H979. [Google Scholar] [CrossRef]
- Li, Y.; Cai, X.; Guan, Y.; Wang, L.; Wang, S.; Li, Y.; Fu, Y.; Gao, X.; Su, G. Adiponectin Upregulates MiR-133a in Cardiac Hypertrophy through AMPK Activation and Reduced ERK1/2 Phosphorylation. PLoS ONE 2016, 11, e0148482. [Google Scholar] [CrossRef] [PubMed]
- Ceylan-Isik, A.F.; Kandadi, M.R.; Xu, X.; Hua, Y.; Chicco, A.J.; Ren, J.; Nair, S. Apelin administration ameliorates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J. Mol. Cell. Cardiol. 2013, 63, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.S.; Zheng, H.; Sharma, N.M.; Shahshahan, H.R.; Patel, K.P.; Mishra, P.K. Lack of miR-133a Decreases Contractility of Diabetic Hearts: A Role for Novel Cross Talk Between Tyrosine Aminotransferase and Tyrosine Hydroxylase. Diabetes 2016, 65, 3075–3090. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.S.; Shahshahan, H.R.; Shang, Q.; Kutty, S.; Boska, M.; Mishra, P.K. MiR-133a Mimic Alleviates T1DM-Induced Systolic Dysfunction in Akita: An MRI-Based Study. Front. Physiol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, L.; Zhang, S.; Zhang, Y.; Lei, S. MicroRNA-1 suppresses cardiac hypertrophy by targeting nuclear factor of activated T cells cytoplasmic 3. Mol. Med. Rep. 2015, 12, 8282–8288. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stølen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Song, X.W.; Zou, J.; Wang, G.K.; Kremneva, E.; Li, X.Q.; Zhu, N.; Sun, T.; Lappalainen, P.; Yuan, W.J.; et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J. Cell Sci. 2010, 123, 2444–2452. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Tang, C.; Zhu, W.; Zhu, J.; Lin, Q.; Fu, Y.; Deng, C.; Xue, Y.; Yang, M.; Wu, S.; et al. CDK6 mediates the effect of attenuation of miR-1 on provoking cardiomyocyte hypertrophy. Mol. Cell. Biochem. 2016, 412, 289–296. [Google Scholar] [CrossRef]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef]
- Li, Q.; Xie, J.; Wang, B.; Li, R.; Bai, J.; Ding, L.; Gu, R.; Wang, L.; Xu, B. Overexpression of microRNA-99a Attenuates Cardiac Hypertrophy. PLoS ONE 2016, 11, e0148480. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Yuan, M.; Zhou, R.; Bai, Q.; Zhang, W.; Zhang, M.; Huang, Y.; Shi, L. MicroRNA-101 inhibits rat cardiac hypertrophy by targeting Rab1a. J. Cardiovasc. Pharmacol. 2015, 65, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Flores, C.; Rivera-Mejías, P.; Vásquez-Trincado, C.; López-Crisosto, C.; Morales, P.E.; Pennanen, C.; Polakovicova, I.; Aliaga-Tobar, V.; García, L.; Roa, J.C.; et al. Angiotensin-(1-9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ. 2020, 27, 2586–2604. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gwathmey, J.K.; Xie, L.H. Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2012, 2, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 2006, 20, 1546–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Santos, C.X.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 73, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Belo, V.A.; Guimarães, D.A.; Castro, M.M. Matrix Metalloproteinase 2 as a Potential Mediator of Vascular Smooth Muscle Cell Migration and Chronic Vascular Remodeling in Hypertension. J. Vasc. Res. 2015, 52, 221–231. [Google Scholar] [CrossRef]
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Strüber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fan, D.; Wang, C.; Wang, J.Y.; Cui, X.B.; Wu, D.; Zhou, Y.; Wu, L.L. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc. Res. 2011, 91, 80–89. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.R.; Shamhart, P.E.; Naugle, J.E.; Meszaros, J.G. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 2008, 51, 704–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Omura, T.; Yoshiyama, M.; Kim, S.; Matsumoto, R.; Nakamura, Y.; Izumi, Y.; Ichijo, H.; Sudo, T.; Akioka, K.; Iwao, H.; et al. Involvement of apoptosis signal-regulating kinase-1 on angiotensin II-induced monocyte chemoattractant protein-1 expression. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Balakumar, P.; Jagadeesh, G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell. Signal. 2014, 26, 2147–2160. [Google Scholar] [CrossRef]
- Ongherth, A.; Pasch, S.; Wuertz, C.M.; Nowak, K.; Kittana, N.; Weis, C.A.; Jatho, A.; Vettel, C.; Tiburcy, M.; Toischer, K.; et al. p63RhoGEF regulates auto- and paracrine signaling in cardiac fibroblasts. J. Mol. Cell. Cardiol. 2015, 88, 39–54. [Google Scholar] [CrossRef]
- Li, C.; Zhen, G.; Chai, Y.; Xie, L.; Crane, J.L.; Farber, E.; Farber, C.R.; Luo, X.; Gao, P.; Cao, X.; et al. RhoA determines lineage fate of mesenchymal stem cells by modulating CTGF-VEGF complex in extracellular matrix. Nat. Commun. 2016, 7, 11455. [Google Scholar] [CrossRef]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J. Cell Physiol. 2016, 231, 1130–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriguchi, Y.; Matsubara, H.; Mori, Y.; Murasawa, S.; Masaki, H.; Maruyama, K.; Tsutsumi, Y.; Shibasaki, Y.; Tanaka, Y.; Nakajima, T.; et al. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ. Res. 1999, 84, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Tian, X.; Qian, Y.; Skibba, M.; Zou, C.; Liu, Z.; Wang, J.; Xu, Z.; Li, X.; Liang, G. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J. Cell. Mol. Med. 2016, 20, 482–494. [Google Scholar] [CrossRef]
- Bai, D.; Ge, L.; Gao, Y.; Lu, X.; Wang, H.; Yang, G. Cytoplasmic translocation of HuR contributes to angiotensin II induced cardiac fibrosis. Biochem. Biophys. Res. Commun. 2015, 463, 1273–1277. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [Green Version]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Wenzel, S.; Taimor, G.; Piper, H.M.; Schlüter, K.D. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-beta expression in adult ventricular cardiomyocytes. FASEB J. 2001, 15, 2291–2293. [Google Scholar] [CrossRef]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Wang, B.; Jones, S.C.; Jassal, D.S.; Dixon, I.M. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H3020–H3030. [Google Scholar] [CrossRef]
- Carvajal, G.; Rodríguez-Vita, J.; Rodrigues-Díez, R.; Sánchez-López, E.; Rupérez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Zhu, H.J.; Huang, X.R.; Lai, K.N.; Johnson, R.J.; Lan, H.Y. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J. Am. Soc. Nephrol. 2002, 13, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Omar, A.; Angelovska, T.; Drobic, V.; Rattan, S.G.; Jones, S.C.; Dixon, I.M. Regulation of collagen synthesis by inhibitory Smad7 in cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1282–H1290. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, G.; Hu, M.C.; Yao, Z.; Tan, T.H. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 1997, 272, 22771–22775. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Hussain, S.R.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.H.; Ning, B.; Ma, X.E.; Gong, W.M.; Jia, T.H. Regulatory roles of microRNA-21 in fibrosis through interaction with diverse pathways (Review). Mol. Med. Rep. 2016, 13, 2359–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, O.; Löhfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.L.; Schäfers, H.J.; Böhm, M.; Laufs, U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol 2012, 107, 278. [Google Scholar] [CrossRef] [PubMed]
- Siddesha, J.M.; Valente, A.J.; Yoshida, T.; Sakamuri, S.S.; Delafontaine, P.; Iba, H.; Noda, M.; Chandrasekar, B. Docosahexaenoic acid reverses angiotensin II-induced RECK suppression and cardiac fibroblast migration. Cell. Signal. 2014, 26, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, J.M.; Schauerte, C.; Hübner, A.; Kölling, M.; Martino, F.; Scherf, K.; Batkai, S.; Zimmer, K.; Foinquinos, A.; Kaucsar, T.; et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur. Heart J. 2015, 36, 2184–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Meng, X.M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef] [Green Version]
- García, R.; Nistal, J.F.; Merino, D.; Price, N.L.; Fernández-Hernando, C.; Beaumont, J.; González, A.; Hurlé, M.A.; Villar, A.V. p-SMAD2/3 and DICER promote pre-miR-21 processing during pressure overload-associated myocardial remodeling. Biochim. Biophys. Acta 2015, 1852, 1520–1530. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Gan, H.; Zhang, H.; Tang, W.; Sun, Y.; Tang, X.; Kong, D.; Zhou, J.; Wang, Y.; Zhu, Y. MicroRNA-21 inhibits SMAD7 expression through a target sequence in the 3′ untranslated region and inhibits proliferation of renal tubular epithelial cells. Mol. Med. Rep. 2014, 10, 707–712. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis after Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFβR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.N.; Yu, C.H.; Pan, M.X.; Zhang, Y.; Zheng, B.J.; Yang, Q.J.; Zheng, Z.M.; Meng, Y. Mir-21 Mediates the Inhibitory Effect of Ang (1-7) on AngII-induced NLRP3 Inflammasome Activation by Targeting Spry1 in lung fibroblasts. Sci. Rep. 2017, 7, 14369. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.P.; et al. Crucial Role of miR-433 in Regulating Cardiac Fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Deng, L.; Zhao, D.; Chen, L.; Yao, Z.; Guo, X.; Liu, X.; Lv, L.; Leng, B.; Xu, W.; et al. MicroRNA-503 promotes angiotensin II-induced cardiac fibrosis by targeting Apelin-13. J. Cell. Mol. Med. 2016, 20, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, R.; Zhu, C.; Qiao, H.; Liu, Y.; Ji, H.; Miao, J.; Chen, L.; Liu, X.; Yang, Y. MiR-503 suppresses hypoxia-induced proliferation, migration and angiogenesis of endothelial progenitor cells by targeting Apelin. Peptides 2018, 105, 58–65. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, X.L.; Li, L.B.; Huang, L.Y.; Tang, Z.; Luo, J.; Yang, L.; Qin, A.P.; Hu, F. miR-503/Apelin-12 mediates high glucose-induced microvascular endothelial cells injury via JNK and p38MAPK signaling pathway. Regen. Ther. 2020, 14, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Desai, V.G.; C Kwekel, J.; Vijay, V.; Moland, C.L.; Herman, E.H.; Lee, T.; Han, T.; Lewis, S.M.; Davis, K.J.; Muskhelishvili, L.; et al. Early biomarkers of doxorubicin-induced heart injury in a mouse model. Toxicol. Appl. Pharmacol. 2014, 281, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Piegari, E.; Russo, R.; Cappetta, D.; Esposito, G.; Urbanek, K.; Dell’Aversana, C.; Altucci, L.; Berrino, L.; Rossi, F.; De Angelis, A. MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget 2016, 7, 62312–62326. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Qi, Y.; Du, J.Q.; Zhang, D.F. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef]
- Wei, Y.; Yan, X.; Yan, L.; Hu, F.; Ma, W.; Wang, Y.; Lu, S.; Zeng, Q.; Wang, Z. Inhibition of microRNA-155 ameliorates cardiac fibrosis in the process of angiotensin II-induced cardiac remodeling. Mol. Med. Rep. 2017, 16, 7287–7296. [Google Scholar] [CrossRef]
- Wang, J.; Guo, L.; Shen, D.; Xu, X.; Wang, J.; Han, S.; He, W. The Role of c-SKI in Regulation of TGFβ-Induced Human Cardiac Fibroblast Proliferation and ECM Protein Expression. J. Cell. Biochem. 2017, 118, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Eissa, M.G.; Artlett, C.M. The MicroRNA miR-155 Is Essential in Fibrosis. Noncoding RNA 2019, 5, E23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Kim, I.K.; Kumar, S.; Jayasinghe, S.; Hong, N.; Castoldi, G.; Catalucci, D.; Jones, W.K.; Gupta, S. NF-κB mediated miR-26a regulation in cardiac fibrosis. J. Cell Physiol. 2013, 228, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Li, Z.; Mericskay, M.; Decaux, J.F. Regulation of Connective Tissue Growth Factor and Cardiac Fibrosis by an SRF/MicroRNA-133a Axis. PLoS ONE 2015, 10, e0139858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Wang, F.; Gao, R.; Wu, J.; Ou, Y.; Chen, X.; Wang, T.; Zhou, X.; Zhu, W.; Li, P.; et al. Autophagy inhibition of hsa-miR-19a-3p/19b-3p by targeting TGF-β R II during TGF-β1-induced fibrogenesis in human cardiac fibroblasts. Sci. Rep. 2016, 6, 24747. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Ye, L.; Han, Z.; Liu, Y.; Yang, Y.; Peng, Z.; Fan, J. miR-19b-3p promotes colon cancer proliferation and oxaliplatin-based chemoresistance by targeting SMAD4: Validation by bioinformatics and experimental analyses. J. Exp. Clin. Cancer Res. 2017, 36, 131. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Taylor, N.E.; Lu, L.; Usa, K.; Cowley, A.W.; Ferreri, N.R.; Yeo, N.C.; Liang, M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension 2010, 55, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; van Rooij, E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Cao, H.; Wang, Q.; Ye, J.; Sui, L.; Feng, J.; Cai, X.; Song, H.; Zhang, X.; Chen, X. MiR-22 may Suppress Fibrogenesis by Targeting TGFβR I in Cardiac Fibroblasts. Cell. Physiol. Biochem. 2016, 40, 1345–1353. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.X.; Li, Y.L.; Zhang, C.C.; Zhou, C.Y.; Wang, L.; Xia, Y.L.; Du, J.; Li, H.H. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 2015, 66, 776–785. [Google Scholar] [CrossRef]
- Marques, F.Z.; Campain, A.E.; Tomaszewski, M.; Zukowska-Szczechowska, E.; Yang, Y.H.; Charchar, F.J.; Morris, B.J. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension 2011, 58, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.L.; Marques, F.Z.; Watson, A.M.; Palma-Rigo, K.; Nguyen-Huu, T.P.; Morris, B.J.; Charchar, F.J.; Davern, P.J.; Head, G.A. A novel interaction between sympathetic overactivity and aberrant regulation of renin by miR-181a in BPH/2J genetically hypertensive mice. Hypertension 2013, 62, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.L.; Gueguen, C.; Lim, K.; Eikelis, N.; Stevenson, E.R.; Charchar, F.J.; Lambert, G.W.; Burke, S.L.; Paterson, M.R.; Marques, F.Z.; et al. Neural suppression of miRNA-181a in the kidney elevates renin expression and exacerbates hypertension in Schlager mice. Hypertens. Res. 2020, 43, 1152–1164. [Google Scholar] [CrossRef]
- Satoh, M.; Takahashi, Y.; Tabuchi, T.; Tamada, M.; Takahashi, K.; Itoh, T.; Morino, Y.; Nakamura, M. Circulating Toll-like receptor 4-responsive microRNA panel in patients with coronary artery disease: Results from prospective and randomized study of treatment with renin-angiotensin system blockade. Clin. Sci. (Lond.) 2015, 128, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Krüger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef] [Green Version]
- Goyal, R.; Goyal, D.; Leitzke, A.; Gheorghe, C.P.; Longo, L.D. Brain renin-angiotensin system: Fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod. Sci. 2010, 17, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Song, J.T.; Qu, H.Y.; Bi, C.L.; Huang, X.Z.; Liu, X.X.; Zhang, M. Mechanical stretch suppresses microRNA-145 expression by activating extracellular signal-regulated kinase 1/2 and upregulating angiotensin-converting enzyme to alter vascular smooth muscle cell phenotype. PLoS ONE 2014, 9, e96338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontaraki, J.E.; Marketou, M.E.; Zacharis, E.A.; Parthenakis, F.I.; Vardas, P.E. Differential expression of vascular smooth muscle-modulating microRNAs in human peripheral blood mononuclear cells: Novel targets in essential hypertension. J. Hum. Hypertens. 2014, 28, 510–516. [Google Scholar] [CrossRef]
- Lambert, D.W.; Lambert, L.A.; Clarke, N.E.; Hooper, N.M.; Porter, K.E.; Turner, A.J. Angiotensin-converting enzyme 2 is subject to post-transcriptional regulation by miR-421. Clin. Sci. (Lond.) 2014, 127, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Wang, B.; Zhang, X.F.; Ma, Y.P.; Liu, J.D.; Wang, X.Z. Contribution of renin-angiotensin system to exercise-induced attenuation of aortic remodeling and improvement of endothelial function in spontaneously hypertensive rats. Cardiovasc. Pathol. 2014, 23, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.R.; Unal, H.; Desnoyer, R.; Yue, H.; Bhatnagar, A.; Karnik, S.S. Angiotensin II-regulated microRNA 483-3p directly targets multiple components of the renin-angiotensin system. J. Mol. Cell. Cardiol. 2014, 75, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharjan, S.; Mopidevi, B.; Kaw, M.K.; Puri, N.; Kumar, A. Human aldosterone synthase gene polymorphism promotes miRNA binding and regulates gene expression. Physiol. Genom. 2014, 46, 860–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.F.; Zhang, Y.; Liu, C.X.; Huang, J.; Ding, G.H. microRNA-125b contributes to high glucose-induced reactive oxygen species generation and apoptosis in HK-2 renal tubular epithelial cells by targeting angiotensin-converting enzyme 2. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4055–4062. [Google Scholar] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamcova, M.; Kawano, I.; Simko, F. The Impact of microRNAs in Renin–Angiotensin-System-Induced Cardiac Remodelling. Int. J. Mol. Sci. 2021, 22, 4762. https://doi.org/10.3390/ijms22094762
Adamcova M, Kawano I, Simko F. The Impact of microRNAs in Renin–Angiotensin-System-Induced Cardiac Remodelling. International Journal of Molecular Sciences. 2021; 22(9):4762. https://doi.org/10.3390/ijms22094762
Chicago/Turabian StyleAdamcova, Michaela, Ippei Kawano, and Fedor Simko. 2021. "The Impact of microRNAs in Renin–Angiotensin-System-Induced Cardiac Remodelling" International Journal of Molecular Sciences 22, no. 9: 4762. https://doi.org/10.3390/ijms22094762
APA StyleAdamcova, M., Kawano, I., & Simko, F. (2021). The Impact of microRNAs in Renin–Angiotensin-System-Induced Cardiac Remodelling. International Journal of Molecular Sciences, 22(9), 4762. https://doi.org/10.3390/ijms22094762