
DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes


Abstract
:1. Introduction
2. Results
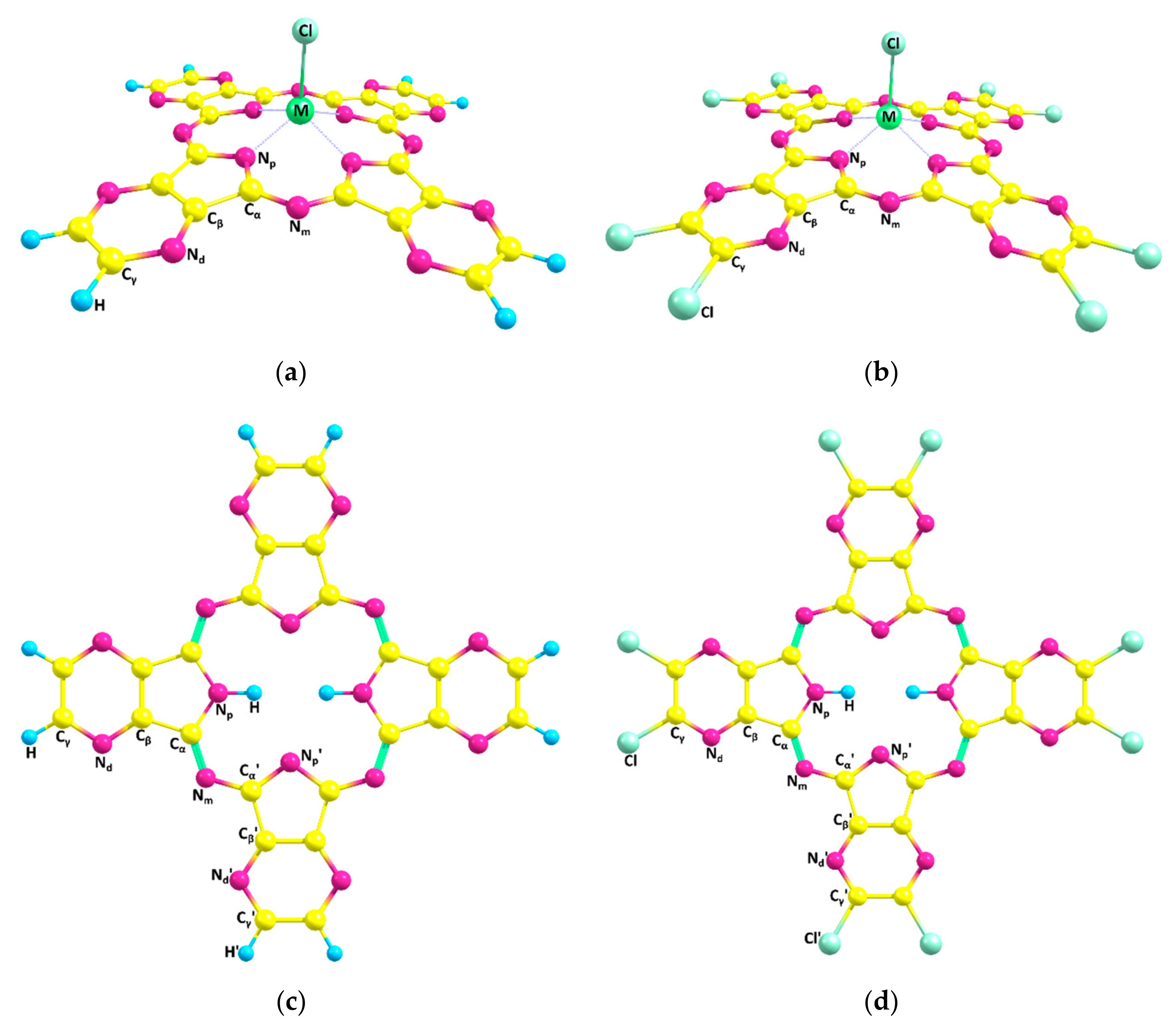
2.1. Molecular Structures
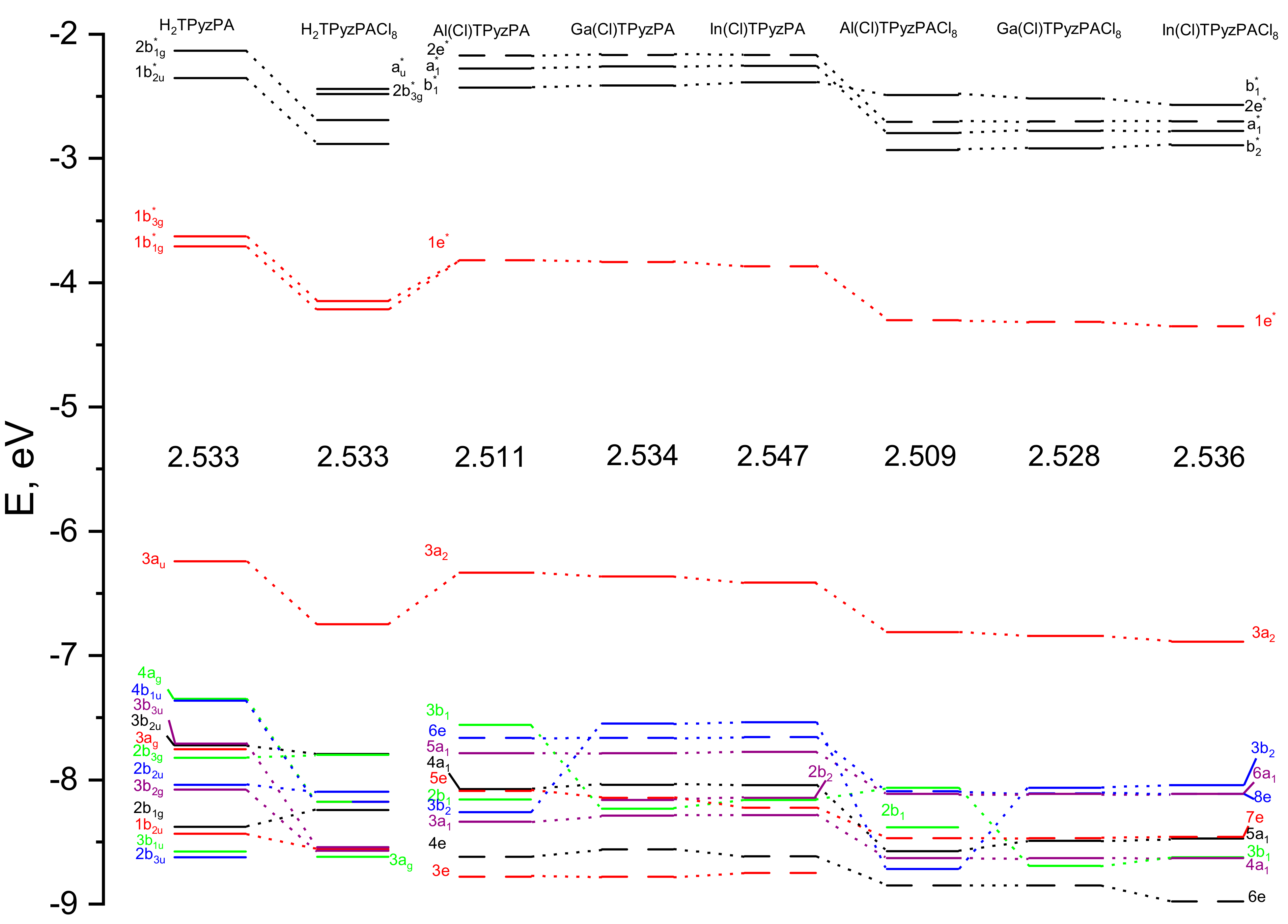
2.2. NBO-Analysis
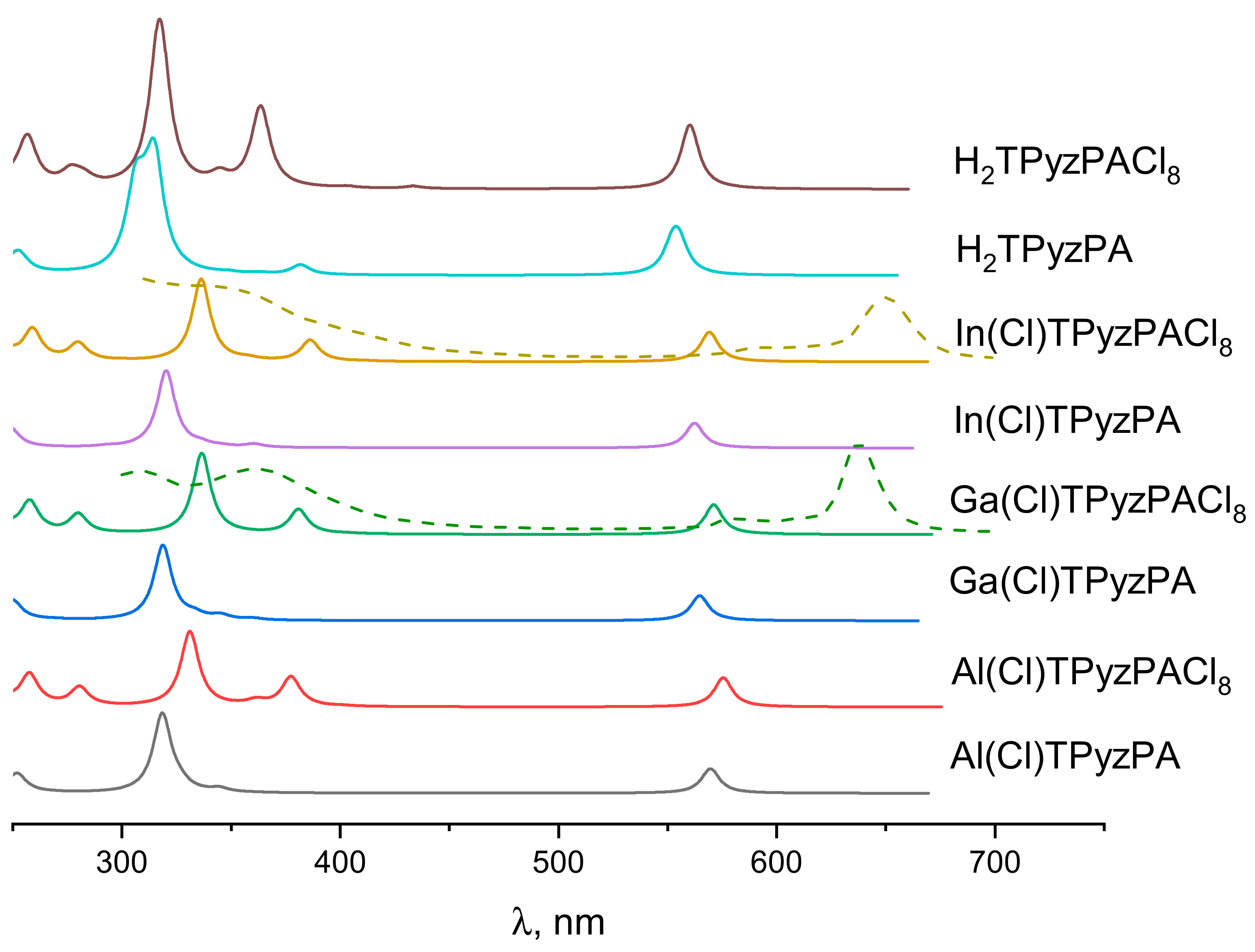
2.3. Electronic Absorption Spectra
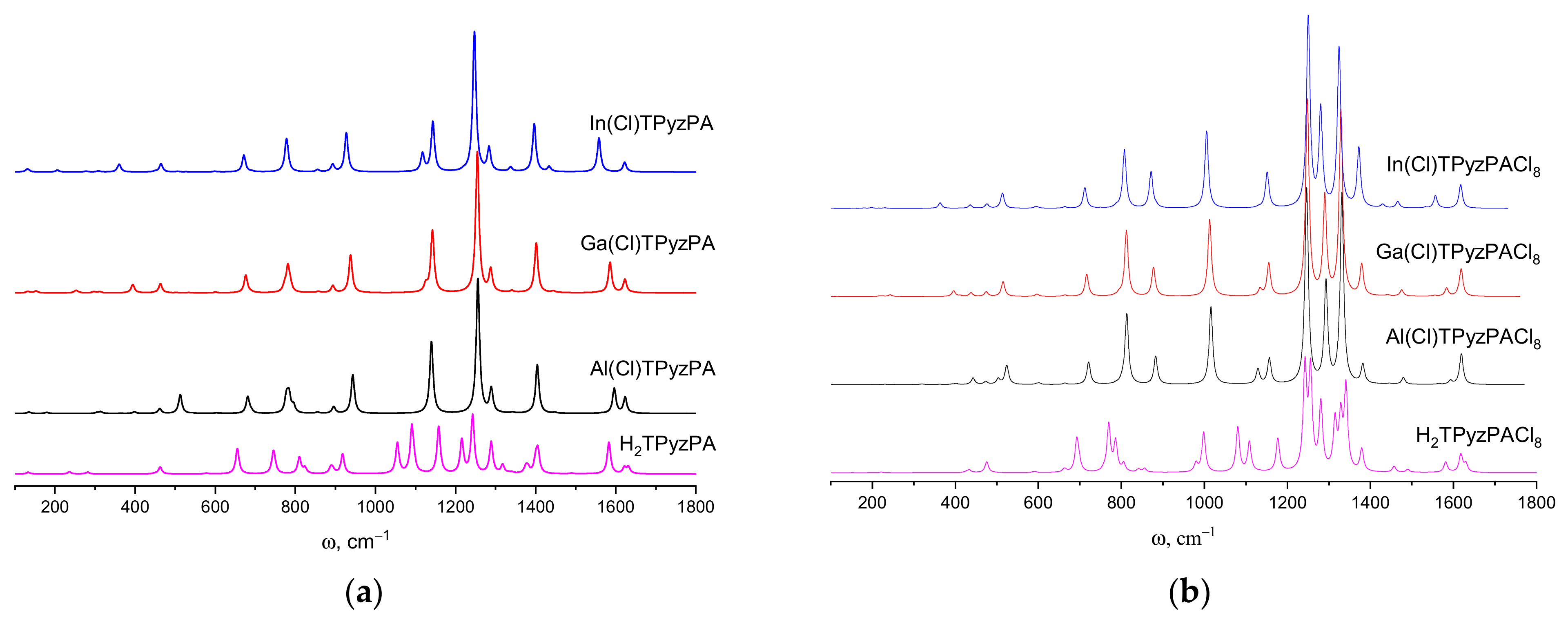
2.4. IR Spectra
3. Materials and Methods
3.1. Synthesis
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De la Torre, G.; Vázquez, P.; Agulló-López, F.; Torres, T. Role of structural factors in the nonlinear optical properties of phthalocyanines and related compounds. Chem. Rev. 2004, 104, 3723–3750. [Google Scholar] [CrossRef] [PubMed]
- Drobizhev, M.; Makarov, N.S.; Rebane, A.; De La Torre, G.; Torres, T. Strong two-photon absorption in Push-Pull phthalocyanines: Role of resonance enhancement and permanent dipole moment change upon excitation. J. Phys. Chem. C 2008, 112, 848–859. [Google Scholar] [CrossRef]
- Senge, M.O.; Fazekas, M.; Notaras, E.G.A.; Blau, W.J.; Zawadzka, M.; Locos, O.B.; Mhuircheartaigh, E.M.N. Nonlinear optical properties of porphyrins. Adv. Mater. 2007, 19, 2737–2774. [Google Scholar] [CrossRef]
- McEwan, K.; Lewis, K.; Yang, G.Y.; Chng, L.L.; Lee, Y.W.; Lau, W.P.; Lai, K.S. Synthesis, Characterization, and Nonlinear Optical Study of Metalloporphyrins. Adv. Funct. Mater. 2003, 13, 863–867. [Google Scholar] [CrossRef]
- Marder, S.R. Organic nonlinear optical materials: Where we have been and where we are going. Chem. Commun. 2006, 2, 131–134. [Google Scholar] [CrossRef]
- Liu, F.; Qin, G.; Li, Z.; Wang, Z.; Peng, M.; Wu, S.; Li, C.; Yang, Y. The design and synthesis of nonlinear optical chromophores containing two short chromophores for an enhanced electro-optic activity†. Mater. Adv. 2021, 2, 728. [Google Scholar] [CrossRef]
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Martynov, A.G.; et al. Macroheterocyclic compounds—A key building block in new functional materials and molecular devices. Macroheterocycles 2020, 13, 311–467. [Google Scholar] [CrossRef]
- Hamdoush, M.; Ivanova, S.S.; Koifman, O.I.; Kos’kina, M.; Pakhomov, G.L.; Stuzhin, P.A. Synthesis, spectral and electrochemical study of perchlorinated tetrapyrazinoporphyrazine and its AlIII, GaIII and InIII complexes. Inorg. Chim. Acta 2016, 444, 81–86. [Google Scholar] [CrossRef]
- Kosov, A.D.; Dubinina, T.V.; Borisova, N.E.; Ivanov, A.V.; Drozdov, K.A.; Trashin, S.A.; De Wael, K.; Kotova, M.S.; Tomilova, L.G. Novel phenyl-substituted pyrazinoporphyrazine complexes of rare-earth elements: Optimized synthetic protocols and physicochemical properties. New J. Chem. 2019, 43, 3153–3161. [Google Scholar] [CrossRef]
- Novakova, V.; Donzello, M.P.; Ercolani, C.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part II: Electronic structure, electrochemical, spectral, photophysical and other application related properties. Coord. Chem. Rev. 2018, 361, 1–73. [Google Scholar] [CrossRef]
- Konovalov, A.I.; Antipin, I.S.; Burilov, V.A.; Madzhidov, T.I.; Kurbangalieva, A.R.; Nemtarev, A.V.; Solovieva, S.E.; Stoikov, I.I.; Mamedov, V.A.; Zakharova, L.Y.; et al. Modern Trends of Organic Chemistry in Russian Universities. Russ. J. Org. Chem. 2018, 54, 157–371. [Google Scholar] [CrossRef]
- Skvortsov, I.A.; Kovkova, U.P.; Zhabanov, Y.A.; Khodov, I.A.; Somov, N.V.; Pakhomov, G.L.; Stuzhin, P.A. Subphthalocyanine-type dye with enhanced electron affinity: Effect of combined azasubstitution and peripheral chlorination. Dyes Pigments 2021, 185, 108944. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Novakova, V.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information. Coord. Chem. Rev. 2016, 309, 107–179. [Google Scholar] [CrossRef]
- Ustimenko, K.A.; Konarev, D.V.; Khasanov, S.S.; Lyubovskaya, R.N. Synthesis of iron(II) octachlorotetrapyrazinoporphyrazine, molecular structure and optical properties of the (X-)2FeTPyzPACl8 dianions with two axial anionic ligands (X- = CN-, CL-). Macroheterocycles 2013, 6, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Faraonov, M.A.; Romanenko, N.R.; Kuzmin, A.V.; Konarev, D.V.; Khasanov, S.S.; Lyubovskaya, R.N. Crystalline salts of the ring-reduced tin(IV) dichloride hexadecachlorophthalocyanine and octachloro- and octacyanotetrapyrazinoporphyrazine macrocycles with strong electron-withdrawing ability. Dyes Pigments 2020, 180, 108429. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Otlyotov, A.A.; Kuzmin, I.A.; Stuzhin, P.A.; Zhabanov, Y.A. DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In. Int. J. Mol. Sci. 2022, 23, 939. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Yablokova, L.I.A.; Ivanova, S.S.; Novakova, V.; Zhabanov, Y.A.; Stuzhin, P.A. Perfluorinated porphyrazines. 3. Synthesis, spectral-luminescence and electrochemical properties of perfluorinated octaphenylporphyrazinatozinc(II). J. Fluor. Chem. 2018, 214, 86–93. [Google Scholar]
- Stuzhin, P.A.; Skvortsov, I.A.; Zhabanov, Y.A.; Somov, N.V.; Razgonyaev, O.V.; Nikitin, I.A.; Koifman, O.I. Subphthalocyanine azaanalogues—Boron(III) subporphyrazines with fused pyrazine fragments. Dyes Pigments 2019, 162, 888–897. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Zhabanov, Y.A.; Pogonin, A.E.; Kuznetsova, A.S.; Islyaikin, M.K.; Girichev, G.V. Gas-phase structures of hemiporphyrazine and dicarbahemiporphyrazine: Key role of interactions inside coordination cavity. J. Mol. Struct. 2019, 1184, 576–582. [Google Scholar] [CrossRef]
- Hamdoush, M.; Nikitin, K.; Skvortsov, I.; Somov, N.; Zhabanov, Y.; Stuzhin, P.A. Influence of heteroatom substitution in benzene rings on structural features and spectral properties of subphthalocyanine dyes. Dyes Pigments 2019, 170, 107584. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Otlyotov, A.A.; Zhabanov, Y.A.; Veretennikov, V.V.; Islyaikin, M.K. Complexes of Ca(II), Ni(II) and Zn(II) with hemi- and dicarbahemiporphyrazines: Molecular structure and features of metal-ligand bonding. Macroheterocycles 2021, 14, 119–129. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Eroshin, A.V.; Stuzhin, P.A.; Ryzhov, I.V.; Kuzmin, I.A.; Finogenov, D.N. Molecular structure, thermodynamic and spectral characteristics of metal-free and nickel complex of tetrakis(1,2,5-thiadiazolo)porphyrazine. Molecules 2021, 26, 2945. [Google Scholar] [CrossRef]
- Konarev, D.V.; Faraonov, M.A.; Kuzmin, A.V.; Osipov, N.G.; Khasanov, S.S.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Molecular structures, and optical and magnetic properties of free-base tetrapyrazinoporphyrazine in various reduction states. New J. Chem. 2019, 43, 19214–19222. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, X.; Zhang, Y.; Jiang, J. The molecular, electronic structures and IR and Raman spectra of metal-free, N,N′-dideuterio, and magnesium tetra-2,3-pyrazino-porphyrazines: Density functional calculations. Vib. Spectrosc. 2007, 43, 447–459. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Ryzhov, I.V.; Kuzmin, I.A.; Eroshin, A.V.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules 2020, 26, 113. [Google Scholar] [CrossRef]
- Allred, A.L.; Rochow, E.G. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem. 1958, 5, 264–268. [Google Scholar] [CrossRef]
- Mann, J.B.; Meek, T.L.; Allen, L.C. Configuration energies of the main group elements. J. Am. Chem. Soc. 2000, 122, 2780–2783. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins. Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Weiss, C.; Kobayashi, H.; Gouterman, M. Spectra of porphyrins. Part III. Self-consistent molecular orbital calculations of porphyrin and related ring systems. J. Mol. Spectrosc. 1965, 16, 415–450. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Hadt, R.G.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Influence of molecular geometry, exchange-correlation functional, and solvent effects in the modeling of vertical excitation energies in phthalocyanines using time-dependent density functional theory (TDDFT) and polarized continuum model TDDFT methods: Ca. J. Phys. Chem. A 2007, 111, 12901–12913. [Google Scholar] [CrossRef] [Green Version]
- Belosludov, V.R.; Nevonen, D.; Rhoda, M.H.; Sabin, R.J.; Nemykin, N.V. Simultaneous Prediction of the Energies of Qx and Qy Bands and Intramolecular Charge-Transfer Transitions in Benzoannulated and Non-Peripherally Substituted Metal-Free Phthalocyanines and Their Analogues: No Standard TDDFT Silver Bullet Yet. J. Phys. Chem. A 2018, 123, 132–152. [Google Scholar] [CrossRef]
- Stillman, M.; Mack, J.; Kobayashi, N. Theoretical aspects of the spectroscopy of porphyrins and phthalocyanines. J. Porphyr. Phthalocyanines 2002, 6, 296–300. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. [Google Scholar] [CrossRef]
- Dini, D.; Hanack, M.; Meneghetti, M. Nonlinear optical properties of tetrapyrazinoporphyrazinato indium chloride complexes due to excited-state absorption processes. J. Phys. Chem. B 2005, 109, 12691–12696. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 15 April 2021).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Zhurko, G.A.; Zhurko, D.A. ChemCraft, Version 1.6 (Build 312); Ed. Available online: http://www.chemcraftprog.com/index.html (accessed on 1 December 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Al(Cl)TPyzPA | Ga(Cl)TPyzPA | In(Cl)TPyzPA | H2TPyzPA | |
|---|---|---|---|---|
| Symmetry | C4v | C4v | C4v | D2h |
| Distances | ||||
| M-Np/M-Np′ | 1.981 | 2.022 | 2.161 | - |
| M-Clax | 2.145 | 2.187 | 2.336 | - |
| Np-Cα/Np′-Cα′ | 1.371 | 1.367 | 1.364 | 1.368/1.355 |
| Cα-Nm/Cα′-Nm | 1.308 | 1.310 | 1.316 | 1.304/1.321 |
| Cα-Cβ/Cα′-Cβ′ | 1.450 | 1.453 | 1.458 | 1.450/1.464 |
| Cβ-Cβ/Cβ′-Cβ′ | 1.391 | 1.393 | 1.399 | 1.401/1.393 |
| Cβ-Nd/Cβ′-Nd′ | 1.328 | 1.328 | 1.326 | 1.329/1.324 |
| Nd-Cγ/Nd′-Cγ′ | 1.322 | 1.322 | 1.323 | 1.320/1.327 |
| Cγ-Cγ/Cγ′-Cγ′ | 1.408 | 1.407 | 1.405 | 1.408/1.401 |
| Cγ-H/Cγ′-H’ | 1.086 | 1.086 | 1.086 | 1.086/1.086 |
| (Np…Np)opp | 3.859 | 3.918 | 4.037 | 4.054/3.899 |
| (Np…Np)adj | 2.728 | 2.770 | 2.854 | 2.812 |
| P 1 | 21.432 | 21.416 | 21.440 | 21.376 |
| Bond angles | ||||
| (ClaxMNp) | 103.1 | 104.3 | 110.9 | - |
| (MNpCα) | 125.2 | 124.5 | 123.6 | 123.2 |
| (NpCαNm)/(Np′Cα′Nm) | 127.9 | 128.0 | 128.0 | 128.5/128.1 |
| (CαNmCα′) | 122.5 | 123.4 | 125.5 | 124.2 |
| (CαCβNd)/(Cα′Cβ′Nd′) | 129.8 | 129.8 | 129.8 | 129.0/130.6 |
| (CβNdCγ)/(Cβ′Nd′Cγ′) | 113.2 | 113.3 | 113.6 | 113.6/113.3 |
| M-X 2 | 0.448 | 0.499 | 0.772 | - |
| α 3 | 175.5 | 174.3 | 164.7 | 180 |
| Al(Cl)TPyzPACl8 | Ga(Cl)TPyzPACl8 | In(Cl)TPyzPACl8 | H2TPyzPACl8 | |
| Symmetry | C4v | C4v | C4v | D2h |
| Distances | ||||
| M-Np/M-Np′ | 1.980 | 2.021 | 2.162 | - |
| M-Clax | 2.142 | 2.183 | 2.332 | - |
| Np-Cα/Np′-Cα′ | 1.372 | 1.368 | 1.364 | 1.368/1.356 |
| Cα-Nm/Cα′-Nm | 1.308 | 1.310 | 1.316 | 1.304/1.321 |
| Cα-Cβ/Cα′-Cβ′ | 1.447 | 1.450 | 1.455 | 1.448/1.461 |
| Cβ-Cβ/Cβ′-Cβ′ | 1.384 | 1.387 | 1.392 | 1.394/1.386 |
| Cβ-Nd/Cβ′-Nd′ | 1.330 | 1.329 | 1.328 | 1.330/1.325 |
| Nd-Cγ/Nd′-Cγ′ | 1.305 | 1.306 | 1.307 | 1.304/1.310 |
| Cγ-Cγ/Cγ′-Cγ′ | 1.433 | 1.432 | 1.430 | 1.433/1.424 |
| Cγ-Cl/Cγ′-Cl′ | 1.709 | 1.709 | 1.709 | 1.709/1.712 |
| (Np…Np)opp | 3.856 | 3.915 | 4.031 | 4.051/3.892 |
| (Np…Np)adj | 2.726 | 2.768 | 2.850 | 2.809 |
| P 1 | 21.440 | 21.424 | 21.440 | 21.376 |
| Bond angles | ||||
| (ClaxMNp) | 103.2 | 104.5 | 111.2 | - |
| (MNpCα) | 125.2 | 124.5 | 123.5 | 123.3 |
| (NpCαNm)/(Np′Cα′Nm) | 128.0 | 128.1 | 128.1 | 128.5/128.2 |
| (CαNmCα′) | 122.3 | 123.3 | 125.3 | 124.0 |
| (CαCβNd)/(Cα′Cβ′Nd′) | 130.3 | 130.3 | 130.3 | 129.5/131.1 |
| (CβNdCγ)/(Cβ′Nd′Cγ′) | 114.9 | 115.0 | 115.2 | 115.3/114.9 |
| M-X 2 | 0.451 | 0.504 | 0.782 | - |
| A 3 | 175.3 | 174.0 | 164.6 | 180 |
| Al(Cl) TPyzPA | Al(Cl) TPyzPACl8 | Ga(Cl) TPyzPA | Ga(Cl) TPyzPACl8 | In(Cl) TPyzPA | In(Cl) TPyzPACl8 | |
|---|---|---|---|---|---|---|
| E(HOMO),eV | −6.329 | −6.803 | −6.362 | −6.836 | −6.411 | −6.882 |
| E(LUMO),eV | −3.815 | −4.294 | −3.829 | −4.308 | −3.864 | −4.343 |
| ∆E, eV | 2.514 | 2.509 | 2.533 | 2.528 | 2.547 | 2.539 |
| q(M) NPA, e | 1.718 | 1.716 | 1.637 | 1.635 | 1.694 | 1.692 |
| q(N) NPA, e | −0.651 | −0.651 | −0.631 | −0.631 | −0.623 | −0.623 |
| q(Cl) NPA, e | −0.550 | −0.545 | −0.523 | −0.518 | −0.528 | −0.521 |
| configuration | 3s0.423p0.83 | 3s0.423p0.83 | 4s0.534p0.81 | 4s0.534p0.81 | 5s0.545p0.76 | 5s0.545p0.76 |
| ∑ E(d-a), kcal mol−1 | 526.0 | 526.5 | 539.6 | 539.0 | 510.7 | 507.8 |
| Q(M-N), e | 0.335 | 0.330 | 0.343 | 0.342 | 0.327 | 0.325 |
| r(M-N), Å | 1.981 | 1.980 | 2.022 | 2.021 | 2.161 | 2.162 |
| Al(Cl)TPyzPA | Al(Cl)TPyzPACl8 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| State | Composition (%) | λ, nm | f | exp. λ, nm | State | Composition (%) | λ, nm | f | exp. λ, nm |
| 11E | (8) (90) | 570 | 0.31 | 11E | (5) (90) | 576 | 0.37 | 643 | |
| 71E | (54) (17) (22) | 327 | 0.07 | 41E | (5) (77) (7) (5) | 377 | 0.37 | ||
| 91E | (27) (58) (6) (6) | 318 | 1.00 | 81E | (70) (7) (7) (9) | 331 | 0.95 | 363 | |
| Ga(Cl)TPyzPA | Ga(Cl)TPyzPACl8 | ||||||||
| State | Composition (%) | λ, nm | f | exp. λ, nm | State | Composition (%) | λ, nm | f | exp. λ, nm |
| 11E | (8) (90) | 565 | 0.32 | 11E | (6) (90) | 571 | 0.38 | 638 | |
| 71E | (26) (34) (32) | 333 | 0.05 | 41E | (6) (75) (13) | 381 | 0.31 | ||
| 91E | (47) (38) (6) (5) | 319 | 0.95 | 81E | (63) (8) (8) (16) | 337 | 1.03 | 360 | |
| In(Cl)TPyzPA | In(Cl)TPyzPACl8 | ||||||||
| State | Composition (%) | λ, nm | f | exp. λ, nm | State | Composition (%) | λ, nm | f | exp. λ, nm |
| 11E | (8) (89) | 562 | 0.32 | 589 | 11E | (90) (6) | 569 | 0.38 | 649 |
| 71E | (36) (26) (28) (7) | 336 | 0.04 | 335 | 41E | (76) (6) (14) | 386 | 0.26 | |
| 91E | (43) (39) (7) (7) | 320 | 0.98 | 81E | (55) (6) (8) (22) | 336 | 1.05 | 339 | |
| H2TPyzPA | H2 TPyzPACl8 | ||||||||
| State | Composition (%) | λ, nm | f | exp. λ, nm | State | Composition (%) | λ, nm | f | exp. λ, nm |
| 11B1u | (90) | 555 | 0.35 | 645 | 11B1u | (90) | 560 | 0.43 | 656 |
| 11B3u | (86) (5) (8) | 552 | 0.33 | 611 | 11B3u | (5) (7) (86) | 560 | 0.39 | 626 |
| 21B3u | (12) (80) | 382 | 0.12 | 31B3u | (5) (85) (5) | 364 | 0.78 | 358 | |
| 41B3u | (7) (66) (9) (9) (6) | 315 | 1.32 | 330 | 41B1u | (18) (78) | 362 | 0.26 | 358 |
| 41B1u | (62) (33) | 311 | 0.29 | 41B3u | (6) (91) | 345 | 0.13 | ||
| 51B1u | (33) (42) (10) (6) (5) | 306 | 0.94 | 51B3u | (79) (8) | 319 | 0.88 | ||
| 51B1u | (68) (12) | 316 | 1.34 | ||||||
| Frequency, cm−1 | Irel, % | Symmetry | Assignment 1 | Exp, cm−1 |
|---|---|---|---|---|
| H2TPyzPA | KBr [26] | |||
| 1091 (ω86) | 78 | B1u | r(Np-Cα) (29), r(Nm-Cα) (18), r(Nd-Cγ) (10), r(Cγ-Cγ) (12) | 1040 |
| 1157 (ω90) | 81 | B2u | r(Np-Cα) (23), r(Nm-Cα) (18), r(Cα-Cβ) (6), r(Cβ-Nd) (8), r(Nd-Cγ) (6), φ(Cα-Np-Hc) (8), φ(Cβ-Nd-Cγ) (7) | 1121 |
| 1242 (ω95) | 100 | B1u | r(Cα-Cβ) (14), r(Cβ-Cβ) (10), r(Nd-Cγ) (10), φ(Nd-Cγ-Hs) (7)r(Cβ-Nd) (6), r(Cβ-Cβ) (42), r(Cγ-Cl) (26), φ(Nd-Cγ-Cl) (6) | 1198 |
| 3193 (ω141) | 24 | B2u | r(Hs-Cγ) (99) | 3051 |
| 3552 (ω143) | 45 | B1u | r(Hc-Np) (99) | 3286 |
| H2TPyzPACl8 | KBr [8] | |||
| 1242 (ω107) | 100 | B1u | r(Cβ-Nd) (6), r(Cβ-Cβ) (42), r(Cγ-Cl) (26), φ(Nd-Cγ-Cl) (6) | 1192 |
| 1255 (ω109) | 98 | B2u | r(Cβ-Nd) (8), r(Cγ-Cγ) (46), r(Cγ-Cl) (21), φ(Nd-Cγ-Cl) (6) | 1235 |
| 1281 (ω111) | 67 | B1u | r(Cα-Cβ) (8), r(Cβ-Cβ) (19), r(Nd-Cγ) (13), r(Cγ-Cγ) (13) | |
| 1328 (ω116) | 52 | B1u | r(Np-Cα) (7), r(Nm-Cα) (11), r(Cβ-Nd) (12), r(Nd-Cγ) (45) | |
| 1340 (ω117) | 83 | B2u | r(Np-Cα) (8), r(Cα-Cβ) (10), r(Cβ-Cβ) (8), r(Cβ-Nd) (19), φ(Cα-Np-Hc) (28) | |
| 3554 (ω143) | 27 | B1u | r(Hc-Np) (99) | 3287 |
| Al(Cl)TPyzPA | ||||
| 1140 (ω92-ω93) | 53 | E | r(Np-Cα) (24), r(Nm-Cα) (14), r(Cα-Cβ) (10), r(Cβ-Nd) (12), r(Nd-Cγ) (9) | |
| 1255 (ω98-ω99) | 100 | E | r(Np-Cα) (5), r(Cα-Cβ) (15), r(Cβ-Cβ) (14), r(Nd-Cγ) (20), φ(Cβ-Nd-Cγ) (9) | |
| 1404(ω113-ω114) | 36 | E | r(Cβ-Cβ) (12), r(Cγ-Cγ) (13), φ(Nd-Cγ-Hs) (36), φ(Cγ-Cγ-Hs) (23) | |
| 3189 (ω142-ω143) | 18 | E | r(Hs-Np) (99) | |
| Al(Cl)TPyzPACl8 | KBr [8] | |||
| 1246 (ω111-ω112) | 100 | E | r(Cγ-Cγ) (43), r(Cγ-Cl) (25), φ(Nd-Cγ-Cl) (6) | 1168 |
| 1293 (ω115-ω116) | 52 | E | r(Np-Cα) (8), r(Cβ-Cβ) (15), r(Nd-Cγ) (25), r(Cγ-Cγ) (13) | 1260 |
| 1331 (ω117-ω118) | 97 | E | r(Cα-Cβ) (8), r(Cβ-Nd) (23), r(Nd-Cγ) (49) | 1323 |
| Ga(Cl)TPyzPA | ||||
| 1142 (ω92-ω93) | 44 | E | r(Np-Cα) (49), r(Nm-Cα) (11), r(Cα-Cβ) (9), r(Nd-Cγ) (9) | |
| 1254 (ω98-ω99) | 100 | E | r(Np-Cα) (6), r(Cα-Cβ) (12), r(Cβ-Cβ) (11), r(Cβ-Nd) (9), r(Nd-Cγ) (18) | |
| 1401 (ω113-ω114) | 35 | E | r(Cβ-Cβ) (11), r(Cγ-Cγ) (12), φ(Nd-Cγ-Hs) (36), φ(Cγ-Cγ-Hs) (23) | |
| 3194 (ω142-ω143) | 17 | E | r(Hs-Np) (99) | |
| Ga(Cl)TPyzPACl8 | this work | |||
| 1248 (ω111-ω112) | 100 | E | r(Cγ-Cγ) (41), r(Cγ-Cl) (24), φ(Nd-Cγ-Cl) (6) | 1232 |
| 1290 (ω115-ω116) | 50 | E | r(Np-Cα) (6), r(Cα-Cβ) (7), r(Cβ-Cβ) (17), r(Nd-Cγ) (26), r(Cγ-Cγ) (13) | |
| 1328 (ω117-ω118) | 94 | E | r(Cα-Cβ) (8), r(Cβ-Nd) (22), r(Nd-Cγ) (50) | 1349 |
| In(Cl)TPyzPA | KBr [39] | |||
| 1143 (ω92-ω93) | 36 | E | r(Np-Cα) (49), r(Nm-Cα) (10), r(Cα-Cβ) (6), r(Nd-Cγ) (10) | 1100 |
| 1247 (ω98-ω99) | 100 | E | r(Np-Cα) (5), r(Cα-Cβ) (15), r(Cβ-Cβ) (14), r(Nd-Cγ) (20) | 1213 |
| 1396 (ω113-ω114) | 34 | E | r(Cβ-Cβ) (10), r(Cγ-Cγ) (10), φ(Nd-Cγ-Hs) (34), φ(Cγ-Cγ-Hs) (22) | 1364 |
| 3194 (ω142-ω143) | 17 | E | r(Hs-Np) (99) | 3316 |
| In(Cl)TPyzPACl8 | this work | |||
| 1250 (ω111-ω112) | 100 | E | r(Cγ-Cγ) (44), r(Cγ-Cl) (26), φ(Nd-Cγ-Cl) (6) | 1264 |
| 1280 (ω114-ω115) | 50 | E | r(Np-Cα) (5), r(Cα-Cβ) (7), r(Cβ-Cβ) (19), r(Nd-Cγ) (26) | 1323 |
| 1325 (ω117-ω118) | 83 | E | r(Cα-Cβ) (8), r(Cβ-Nd) (23), r(Nd-Cγ) (49) | 1364 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryzhov, I.V.; Eroshin, A.V.; Zhabanov, Y.A.; Finogenov, D.N.; Stuzhin, P.A. DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes. Int. J. Mol. Sci. 2022, 23, 5379. https://doi.org/10.3390/ijms23105379
Ryzhov IV, Eroshin AV, Zhabanov YA, Finogenov DN, Stuzhin PA. DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes. International Journal of Molecular Sciences. 2022; 23(10):5379. https://doi.org/10.3390/ijms23105379
Chicago/Turabian StyleRyzhov, Igor V., Alexey V. Eroshin, Yuriy A. Zhabanov, Daniil N. Finogenov, and Pavel A. Stuzhin. 2022. "DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes" International Journal of Molecular Sciences 23, no. 10: 5379. https://doi.org/10.3390/ijms23105379
APA StyleRyzhov, I. V., Eroshin, A. V., Zhabanov, Y. A., Finogenov, D. N., & Stuzhin, P. A. (2022). DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes. International Journal of Molecular Sciences, 23(10), 5379. https://doi.org/10.3390/ijms23105379